

Panoramica sulla registrazione dei dispositivi medici in Europa
L'EU, composta da 27 Member States, ha sviluppato il Regolamento sui Dispositivi Medici (MDR) 2017/745 e il Regolamento sui Dispositivi Medico-Diagnostici in Vitro (IVDR) 2017/746, che sono stati interamente implementati di recente. Questi regolamenti sono parte integrante del processo di registrazione dei dispositivi medici in Europa e hanno ora sostituito le Direttive. Entrambi i regolamenti consistono in nuovi requisiti aggiuntivi, e queste saranno le procedure di regolamentazione centralizzate che dovranno essere seguite per l'immissione dei dispositivi medici in uno qualsiasi dei 27 paesi. I produttori esteri di dispositivi medici che non hanno una sede fisica in Europa devono nominare un Rappresentante Autorizzato Europeo (EAR) per aiutarli a conformarsi a questi regolamenti.
![]()
Autorità di regolamentazione: Autorità nazionale competente![]()
Regolamento: Regolamenti sui dispositivi medici (MDR) 2017/745, regolamenti sui dispositivi diagnostici in vitro 2017/746![]()
Percorso normativo: Marchio CE seguito da Registrazione/Notifica![]()
Rappresentante autorizzato: Rappresentante Autorizzato Europeo (EAR) per produttori non-UE![]()
Requisito del SGQ: ISO 13485:2016![]()
Valutazione dei dati tecnici: Organismo notificato per la marcatura CE
Classificazione del dispositivo
La classificazione del dispositivo è il primo passo per determinare il percorso normativo per un determinato prodotto. Esistono circa 22 norme di attuazione per i dispositivi medici e sono classificati come
| Classe | Il rischio |
|---|---|
| Classe I | Basso |
| Classe IIa | Moderato |
| Classe IIb | Da moderato a elevato |
| Classe III | Alto |
Allo stesso modo, per gli IVD, sono state implementate circa 7 regole, classificate nelle seguenti quattro categorie
| Classe | Il rischio |
|---|---|
| Classe A | Basso |
| Classe B | Moderato |
| Classe C | Da moderato a elevato |
| Classe D | Alto |
Date le istruzioni specializzate in vigore per le diverse classi, l'identificazione della giusta classe di dispositivi è fondamentale per determinare il percorso normativo.
Rappresentante autorizzato europeo (EAR)
Qualsiasi produttore straniero che intenda lanciare i propri dispositivi nella regione dell'UE è obbligato a nominare un rappresentante autorizzato europeo (EAR) ai sensi dell'articolo 11 dell'EU MDR e dell'IVDR EU MDR .
Registrazione dei dispositivi medici
Per commercializzare i dispositivi medici nell'area geografica dell'UE, ottenere la marcatura CE è obbligatorio. I produttori sono tenuti a identificare e nominare organismi notificati, sottoporsi alla valutazione di conformità e rilasciare la certificazione CE.
La decodifica delle informazioni normative relative alla registrazione o alla notifica di un dispositivo tramite un sistema di registrazione online può risultare difficile senza l'assistenza di un esperto.
Flusso di processo
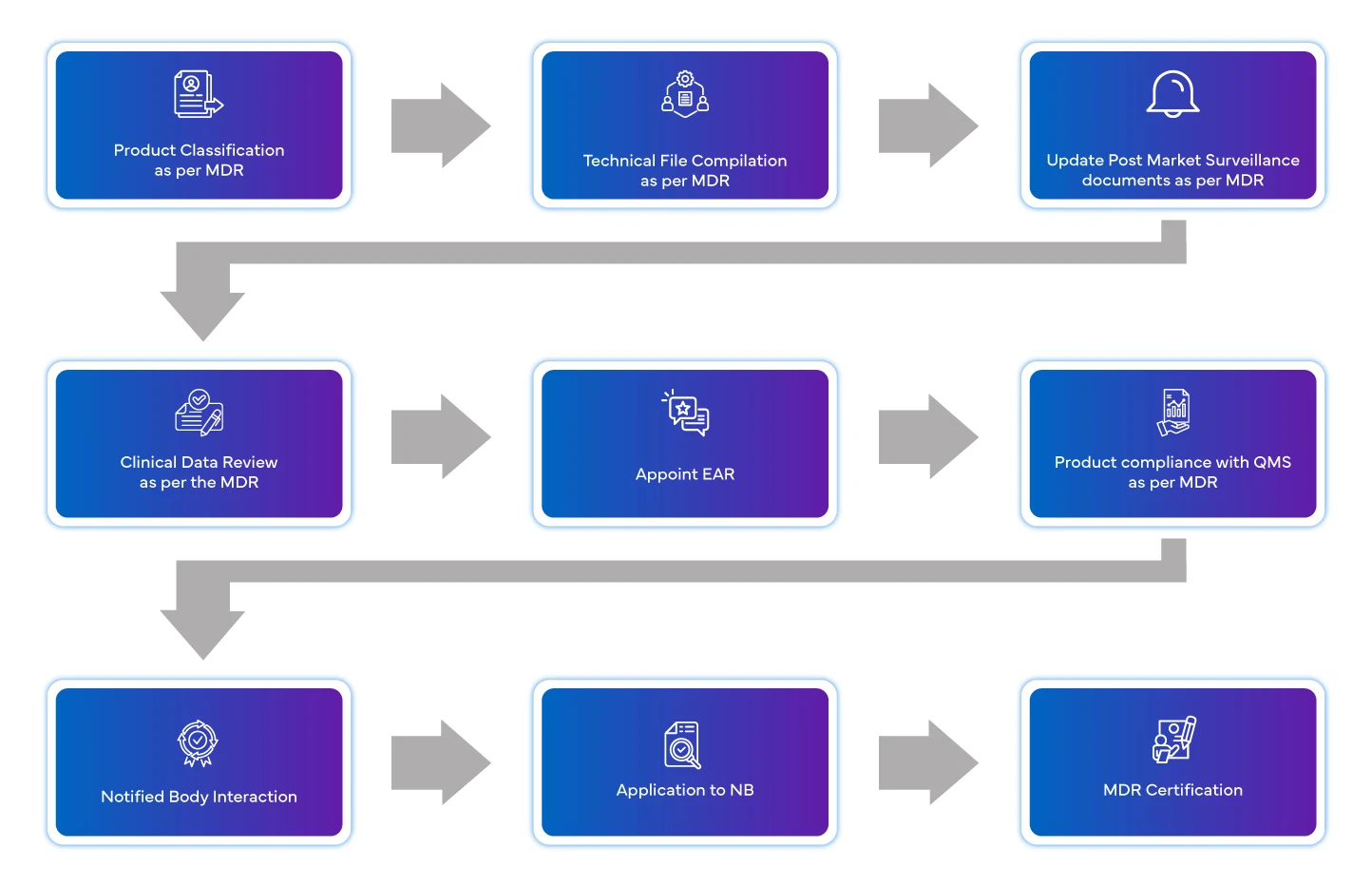
Gestione del ciclo di vita del dispositivo post-approvazione
Le normative europee sui dispositivi medici sottolineano ora l'importanza dei requisiti post-vendita. Il fabbricante è tenuto a dotarsi di un sistema di gestione della sorveglianza. È necessario fornire informazioni periodiche sul dispositivo.
Freyr può supportarvi nella creazione del piano di Post-Market Surveillance (PMS), del rapporto di Post-Market Surveillance (PMSR), del rapporto periodico di aggiornamento sulla sicurezza (PSUR) e del follow-up clinico post-commercializzazione (PMCF)/follow-up delle prestazioni post-commercializzazione (PMPF).
Il supporto di Freyr comprende anche attività come
- Gestione delle modifiche post-approvazione - modifiche alle approvazioni dei dispositivi medici esistenti, come l'aggiunta di nuove varianti e accessori, l'aggiunta di nuove indicazioni d'uso, ecc.
- Mantenimento della certificazione ISO 13485:2016 e CE
- Rinnovo delle licenze
- Collegamento tra l'organismo notificato e il produttore
Registrazione dei dispositivi medici in Europa
Competenza di Freyr
- Classificazione europea dei dispositivi medici
- Supporto del Rappresentante Autorizzato Europeo (EAR)
- ISO 14971:2019 Consultazione sulla gestione del rischio
- Conformità ISO 13485:2016
- Revisione, compilazione e presentazione del fascicolo tecnico/di progetto CE
- Supporto alla transizione EU MDR
- Supporto alla transizione IVDR dell'UE
- Clinical Evaluation Reports (CER) per i dispositivi medici
- Relazioni di valutazione delle prestazioni (PER) per i dispositivi diagnostici in vitro
- Notifica/registrazione di dispositivi medici tramite il sistema di registrazione online
- Rapporto sulla strategia di regolamentazione dei dispositivi medici
- Supporto ai test: biocompatibilità, sicurezza elettrica, meccanica e prestazioni.
- Supporto per la conformità dell'etichettatura
- Supporto GMP
- Supporto per la sorveglianza post-vendita
