Panoramica sulla registrazione dei dispositiviFDA pressoFDA US
Gli Stati Uniti d'America (USA) sono noti per essere un mercato altamente regolamentato per i dispositivi medici, con percorsi e requisiti di registrazione ben definiti. Le prime normative sui dispositivi medici negli Stati Uniti risalgono al 1976 e si sono evolute nel corso del tempo. Sono regolamentate dal Centre for Devices and Radiological Health (CDRH) sotto l'egida della Food and Drug Administration (FDA). Freyr ha aiutato diversi produttori di dispositivi produttori conformarsi al processo di registrazione dei dispositiviFDA US .
![]()
Autorità di regolamentazione: Food and Drug AdministrationFDA)![]()
Regolamento: Titolo 21 Codice dei regolamenti federali (21 CFR) Parti 800 - 1299![]()
Percorso normativo: Notifica pre-commercializzazione o Approvazione pre-commercializzazione o Classificazione De-Novo![]()
Rappresentante autorizzato: Agente statunitense![]()
Requisito del SGQ: Regolamento sul sistema di qualità (QSR) (21 CFR parte 820)![]()
Valutazione dei dati tecnici: Centro per i dispositivi e la salute radiologica![]()
Validità della licenza: Illimitato![]()
Requisiti di etichettatura: 21 CFR Parte 801![]()
Formato di presentazione: Carta e CD/DVD![]()
Lingua: Inglese
Classificazione dei dispositivi medici USA
La FDA classifica i dispositivi medici in 3 categorie basate sul rischio, Classe I, Classe II e Classe III, dove i dispositivi di Classe I sono considerati a basso rischio, mentre quelli di Classe III sono associati ad un rischio elevato. I requisiti e il percorso di registrazione variano a seconda della classe di dispositivi.
| Classe del dispositivo | Il rischio | Percorso di registrazione per l'approvazione |
|---|---|---|
| I | Basso rischio | Esente da 510(k) |
| II | Rischio moderato (Con dispositivo di predicato) | Notifica preliminare all'immissione sul mercato/510(k) |
Rischio moderato (Senza dispositivo di predicato) | Applicazione De-Novo | |
| III | Alto rischio | Approvazione preliminare all'immissione sul mercato (PMA) |
Agente FDA degli Stati Uniti
Le aziende senza uffici locali negli US devono nominare un Agente FDA US per rappresentare il produttore. L'agente FDA US deve risiedere negli US o mantenere una sede di attività negli US. Le responsabilità da adempiere da parte dell'agente sono predeterminate dalla FDA US come parte delle normative CFR.
Naviga attraverso le Domande Frequenti (FAQ) sull'Agente US.
Incontri interattivi con la US FDA
FDA US FDA i produttori diversi tipi di incontriQ-Submissionper raggiungere diversi obiettivi. Tali incontri con l'agenzia prima dell'avvio o durante lo sviluppo del dispositivo, prima della presentazione delle domande di registrazione dei dispositiviFDA US , aiutanoproduttori ottimizzare i tempi e i costi sostenuti per la commercializzazione dei dispositivi.
Registrazione dei dispositivi medici USA
I dispositivi possono essere approvati dal CDRH e dalla FDA attraverso uno dei vari percorsi di registrazione. Sono elencati come:
Dispositivi medici di Classe I: I dispositivi di classe I sono solitamente esenti da GMP e dalla presentazione 510(k) e non richiedono l'approvazione preventiva della US FDA per essere commercializzati negli US. Altri requisiti come la registrazione dello stabilimento, l'elenco dei dispositivi, UDI, PMS ecc. devono essere rispettati dal produttore.
Dispositivi medici di Classe II: I dispositivi a rischio medio con dispositivi predicatori approvati 510(k) possono optare per la Notifica Pre-commercializzazione 510(k) (PMN), anche chiamata registrazione 510(k). Il dispositivo in questione deve stabilire l'Equivalenza Sostanziale (SE) con i dispositivi predicatori identificati e dichiarati. Questo percorso è il più ampiamente adottato per la registrazione dei dispositivi negli US. I produttori di dispositivi a rischio medio senza predicatori possono richiedere la classificazione da parte della US FDA tramite le domande De-Novo.
Dispositivi medici di Classe III: I produttori di dispositivi di Classe III ad alto rischio devono presentare una domanda di Approvazione Pre-commercializzazione (PMA) alla US FDA. I dispositivi devono essere sottoposti a una valutazione clinica dettagliata e il produttore deve presentare dati dettagliati sulla sicurezza e l'efficacia derivanti da studi clinici. La US FDA effettuerà un'ispezione QMS come parte della valutazione prima di rilasciare un' Approvazione Pre-commercializzazione per il dispositivo.
Registrazioni di dispositivi medici non CDRH
In base alle indicazioni d'uso, alcuni prodotti border line considerati come dispositivi medici in altri Paesi, come respiratori chirurgici, disinfettanti, prodotti combinati, coinvolgono altre Agenzie come il Centro per il Controllo delle Malattie (CDC), l'Istituto Nazionale per la Sicurezza e i Rischi sul Lavoro (NIOSH), l'Agenzia per la Protezione dell'AmbienteEPA), il Centro per la Valutazione e la Ricerca Biologica (CBER), il Centro per la Valutazione e la Ricerca sui Farmaci (CDER).
Requisiti di conformità post-approvazione per i dispositivi medici
Tutti i produttori di dispositivi devono rispettare i requisiti post-approvazione elencati di seguito:
- Requisiti per la registrazione e l'inserimento nell'elenco: Gli stabilimenti di tutte le classi di dispositivi devono essere registrati nel database del FURL e il dispositivo deve essere inserito nell'elenco dopo l'ottenimento dell'approvazione e prima della commercializzazione del dispositivo negli Stati Uniti. Alcuni dispositivi, come quelli per le radiazioni, devono soddisfare altri requisiti, come il numero di adesione, prima di poter essere importati negli Stati Uniti.
- Identificazione univoca del dispositivo: Tutte le classi di dispositivi devono essere conformi alle norme sull'identificazione univoca dei dispositivi (UDI) per poterli commercializzare negli Stati Uniti.
- Tasse di stabilimento: Il produttore deve pagare le tasse annuali di stabilimento per mantenere attiva la registrazione del proprio stabilimento e continuare a commercializzare dispositivi negli US. La US FDA ha una struttura tariffaria ridotta per le entità più piccole con un certificato di piccola impresa attivo.
- Audit di qualità: Per i dispositivi non esenti da GMP, la US FDA può ispezionare lo stabilimento di produzione in qualsiasi momento per verificarne la conformità ai Regolamenti sui Sistemi di Qualità (QSR) in conformità con il 21 CFR 820.
Flusso di processo
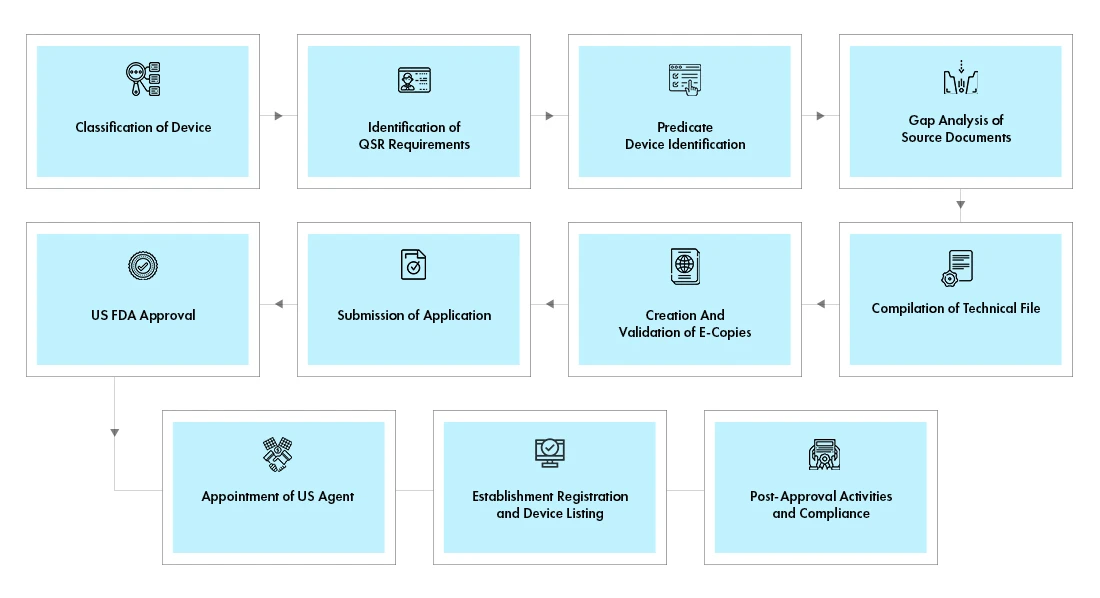
Gestione del ciclo di vita del dispositivo dopo l'approvazione
Freyr supporta i produttori esteri nella gestione End-to-End del ciclo di vita dei dispositivi medici, incluse le attività post-approvazione, come:
- Gestione delle modifiche post-approvazione - modifiche alle approvazioni dei dispositivi medici esistenti, come l'aggiunta di nuove varianti e accessori, l'aggiunta di nuove indicazioni d'uso, ecc.
- Mantenimento delle approvazioni e della registrazione attraverso il pagamento puntuale delle tasse MDUFA alla FDA.
- Collegamento tra la US FDA e il produttore.
Freyr dispone di un centro di distribuzione esclusivo negli US con un team professionale per fornire supporto normativo ai produttori nel mantenimento della qualità e della sicurezza necessarie per l'approvazione. Gli esperti di intelligence di Freyr osservano attentamente gli aggiornamenti normativi e tengono i clienti informati sui passi da intraprendere per la conformità dei prodotti agli standard attuali.
Sintesi
| Il rischio | Classe del dispositivo | Audit del SGQ | Disponibilità del predicato | Percorso normativo | Agente statunitense | US FDA |
|---|---|---|---|---|---|---|
| Basso rischio | I | No | NA | Esente | Sì | 1 mese |
| Rischio medio | II | Sì (post approvazione) | Sì | PMN/510(k) | Sì | 9 - 12 mesi |
| Rischio medio | II | Sì (post approvazione) | No | Richiesta di classificazione de novo | Sì | 18 - 30 mesi |
| Alto rischio | III | Sì (previa approvazione) | NA | PMA | Sì | 18 - 30 mesi |
Servizi di registrazione dei dispositivi medici di Freyr
Competenza di Freyr
- Due diligence normativa
- Documentazione del dispositivo
- 513(g) sostegno
- Registrazione 510(k)
- Richiesta di classificazione de novo
- Registrazione PMA
- 21 CFR 820 conformità
- Supporto all'audit BIMO
- MDSAP Conformità
- Supporto per l'etichettatura
- Supporto alla pubblicazione e alla presentazione
- Agente statunitense
- Riunioni di presentazione Q
- Riunioni RFD e pre-RFD
- Certificazione delle piccole imprese
- Registrazione dello stabilimento ed elenco dei dispositivi
- Conformità normativa per i dispositivi medici a radiazione
- Gestione delle modifiche successive all'approvazione
- Sorveglianza post-mercato
- Conformità UDI
- Consulenza normativa per la risoluzione delle carenze