Panoramica della registrazione del Software as a Medical Device SaMD)
Il Software come Dispositivo Medico (SaMD), chiamato anche Medical Device Software (MDSW), è l'argomento più in voga nel settore sanitario. Il mercato globale dei SaMD è stimato a un valore di 86,5 miliardi di US $ entro il 2027, con un CAGR del 21,9% dal 2020 al 2027. Questa crescita è innescata da vari fattori di conformità dei SaMD, come l'adozione dell'Internet of Things (IoT), delle piattaforme sanitarie digitali e l'adozione di software per il monitoraggio continuo dei parametri fisiologici da parte degli operatori sanitari per l'assistenza remota. Ciò è stato ulteriormente accelerato dalla pandemia di COVID, che ha reso necessari i servizi sanitari remoti e ha a sua volta creato la necessità di un processo semplificato per le registrazioni di Software come Dispositivo Medico (SaMD). Esistono tre tipi di software correlati ai dispositivi medici basati sulla classificazione di Software come Dispositivo Medico (SaMD), quali:
- Software in un dispositivo medico (SiMD)
- Software come dispositivi mediciSaMD)
- Software utilizzato per la produzione/manutenzione di un dispositivo medico
Scenario normativo globale
Per quanto riguarda i SaMD, ne esistono di vari tipi utilizzati per diverse applicazioni, quali screening e diagnosi, monitoraggio e allerta, gestione delle malattie, ecc. Le agenzie sanitarie dei paesi sviluppati o altamente regolamentati, quali UE, US, Canada e Australia, hanno definito normative relative ai SaMD e alcune di esse hanno già elaborato documenti di orientamento, mentre altre sono in fase di elaborazione. Alcuni mercati semi-regolamentati e non regolamentati considerano tali software come dispositivi medici, ma non dispongono di linee guida differenziate e specifiche per la classificazione dei Software as a Medical Device (SaMD). Essi seguono le linee guida armonizzate accettate a livello internazionale per la valutazione e l'approvazione del software. Di seguito sono elencate alcune delle principali linee guida disponibili sui SaMD:
- Guida IMDRF per classificazione, QMS, valutazione della cybersecurity, valutazione clinica
- LEU MDR contiene informazioni dettagliate su questa categoria di dispositivi.
- Guida MDCG sulla qualificazione e classificazione di SaMD, requisiti CER / PER per SaMD
- FDA US in materia di sicurezza informatica, valutazione clinica e requisiti di registrazione per diversi tipi di software quali sistemi decisionali, PACS, applicazioni mobili, ecc.
- Documento guida di Health Canada sulla definizione e la classificazione
- Le nuove norme del TGA per i dispositivi medici basati sul software, che entreranno in vigore dall'agosto 2020.
La registrazione dei SaMD in altri mercati globali deve essere gestita caso per caso e richiede una stretta interazione con la rispettiva agenzia sanitaria per l'approvazione. Il percorso generale seguito per la registrazione dei SaMD comprende:
- Determinare se un dato software si qualifica come SaMD
- Classificazione dei dispositivi in base al rischio
- Identificare gli standard applicabili e i requisiti dei dati da parte dell'Agenzia sanitaria interessata.
- Generare i dati come richiesto dalla rispettiva Agenzia
- Compilazione del fascicolo tecnico in base ai requisiti del paese.
- Presentazione e risoluzione delle domande fino all'approvazione
- Gestione del ciclo di vita post-approvazione
Supporto Normativo End-to-End di Freyr per SaMD
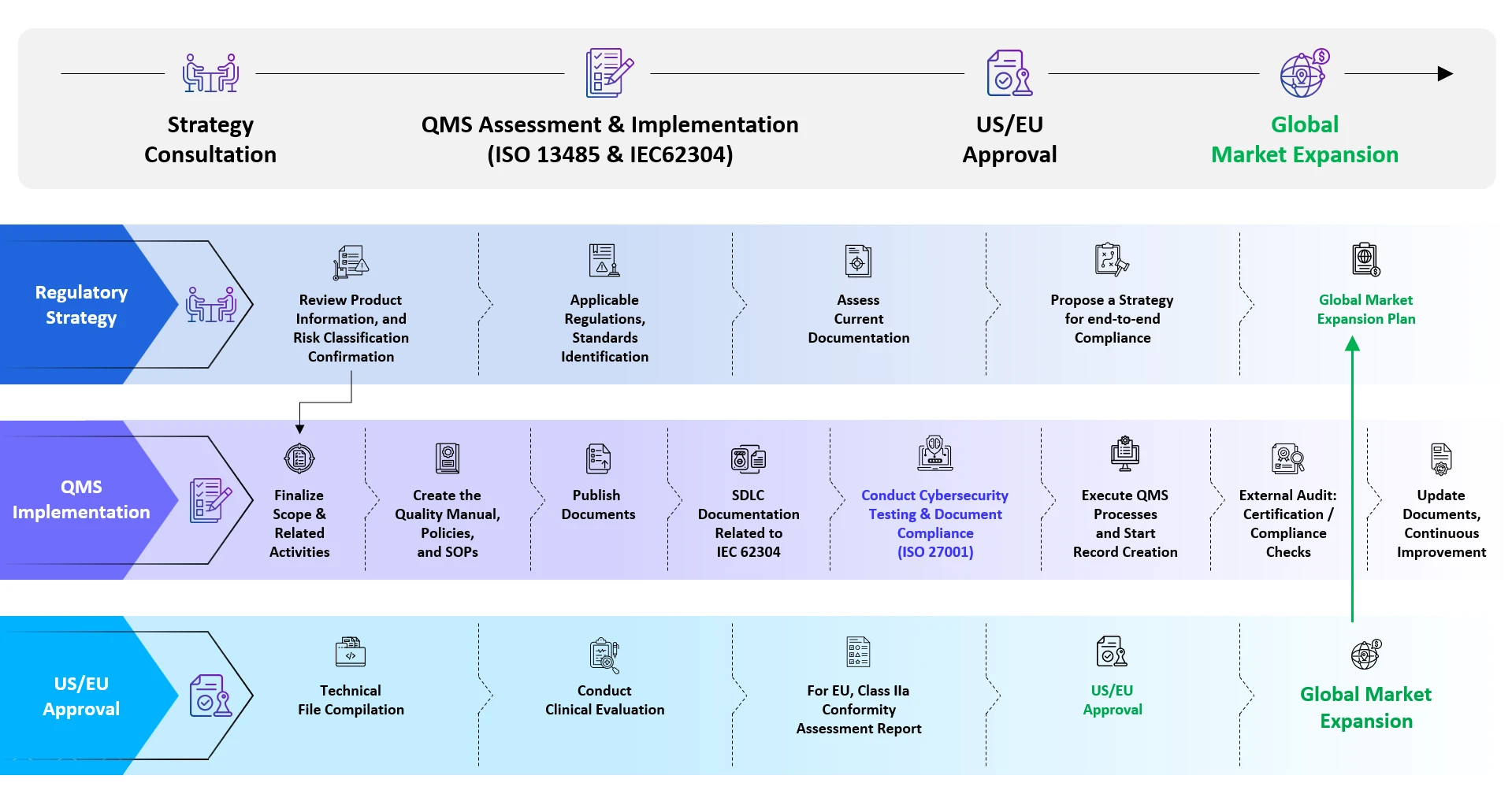
Servizi di registrazione del Software as a Medical Device SaMD)
Registrazione del Software as a Medical Device SaMD) - Competenza
- Regulatory Intelligence Services
- Due diligence normativa/rapporti strategici
- Qualificazione e classificazione di SaMD
- Identificazione degli standard applicabili
- Analisi delle lacune dei documenti di partenza
- Registrazione SaMD
- Conformità Quality Management System (QMS)
- Servizi di consulenza per la valutazione della sicurezza informatica
- Servizi di consulenza su studi di valutazione clinica
- Compilazione di rapporti di valutazione clinica (CER) / rapporti di valutazione del prodotto (PER) ecc.
- Gestione delle modifiche post-approvazione





