Panoramica sul Software as a Medical Device SaMD)
Software as a Medical Device SaMD) è l'ultimo argomento in espansione nel segmento della sanità. Si prevede che il mercato SaMD crescerà a livello globale a un tasso di crescita annuale composto (CAGR) del 10,78%. Questa crescita è innescata da vari fattori, come l'adozione dell'Internet degli oggetti (IoT), le piattaforme sanitarie digitali e l'adozione di software per il monitoraggio continuo dei parametri fisiologici da parte degli operatori sanitari per l'assistenza a distanza. Tuttavia, questo promettente panorama presenta anche sfide uniche, una delle quali è determinare se rientra nella categoria dei dispositivi medici e se aderisce ai requisiti normativi.
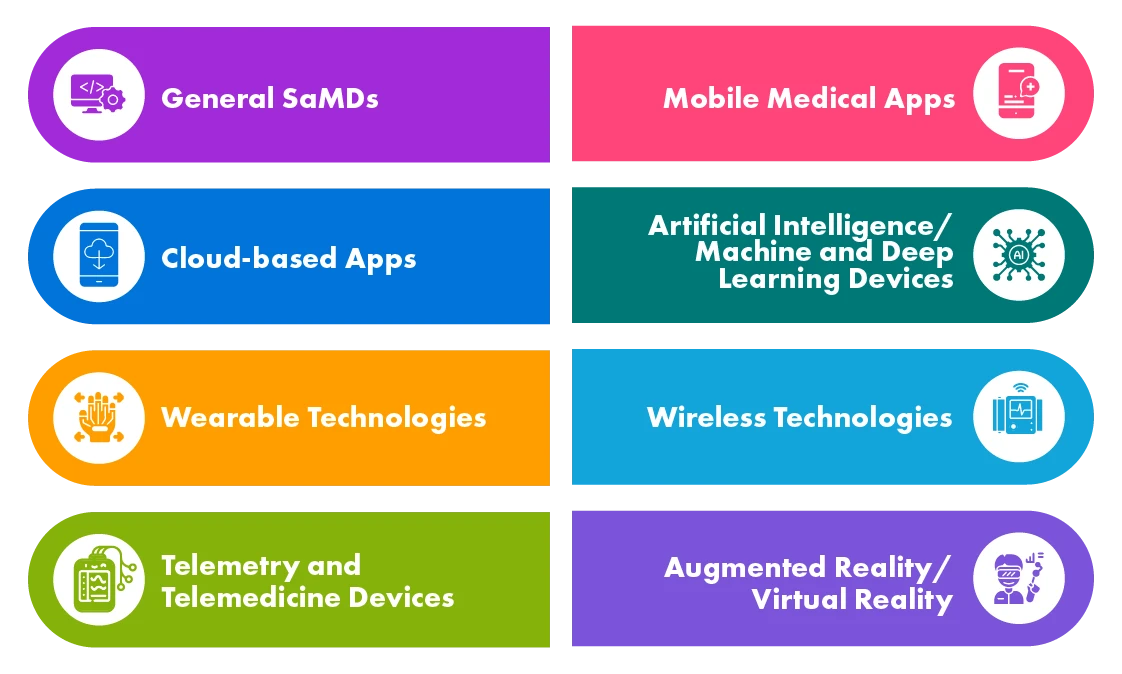
Diversi tipi di prodotti per la salute digitale
Scenario normativo globale per la registrazione del Software as a Medical Device SaMD)
I SaMD sono utilizzati in varie applicazioni come screening e diagnosi, monitoraggio e allerta, gestione delle malattie, ecc. Le Agenzie Sanitarie dei paesi sviluppati come l'UE, gli US, il Canada e l'Australia hanno definito regolamentazioni sui SaMD e alcune di esse hanno già sviluppato documenti guida mentre altre sono in fase di elaborazione.
Alcuni mercati regolamentati e non regolamentati considerano il software come dispositivo medico, ma non hanno linee guida specifiche e differenziate per la classificazione del Software as a Medical Device SaMD). Seguono le linee guida armonizzate accettate a livello internazionale per la valutazione e l'approvazione del software.
Di seguito sono elencate alcune delle principali linee guida disponibili sulla registrazione del Software as a Medical Device SaMD):
- Guida IMDRF per la classificazione, il Quality Management System (QMS), la valutazione della cybersecurity e la valutazione clinica.
- L'EU MDR 2017/745 EU MDR ha definito nel dettaglio i requisiti normativi e le linee guida per questa categoria di dispositivi.
- La guida MDCG sulla qualificazione e la classificazione dei SaMD, i requisiti dei Clinical Evaluation Reports (CER)/Performance Evaluation Report (PER) per i SaMD.
- FDA US in materia di sicurezza informatica, valutazione clinica e requisiti di registrazione per diversi tipi di software, quali sistemi decisionali, sistemi di archiviazione e comunicazione delle immagini (PACS), applicazioni mobili, ecc.
- Documento guida di Health Canada sulla definizione e la classificazione SaMD .
- Le nuove norme del TGA per i dispositivi medici basati su software.
La registrazione dei SaMD in altri mercati globali sarà gestita con un approccio caso per caso e richiede una stretta interazione con la rispettiva Agenzia Sanitaria per l'approvazione. Il percorso generale seguito per la registrazione dei SaMD comprende:
- Determinare se un determinato software si qualifica come SaMD.
- Classificazione dei dispositivi in base al rischio.
- Identificare gli standard applicabili e i dati richiesti dall'Agenzia sanitaria interessata.
- Generare i dati richiesti dalla rispettiva Agenzia.
- Compilazione della documentazione tecnica secondo i requisiti del Paese.
- Presentazione e risoluzione delle domande fino all'approvazione.
- Gestione del ciclo di vita post-approvazione.
Le nostre competenze
- Regulatory Intelligence Services (supporto al mercato e all'etichettatura)
- Due diligence normativa/rapporti strategici
- Qualificazione e classificazione di SaMD
- Classificazione del prodotto Applicazione a NB
- Analisi delle lacune
- Riunioni di pre-presentazione con la FDA
- Identificazione degli standard applicabili
- Attività di gestione del rischio
- Supporto alla gestione del rischio
- Supporto per l'etichettatura
- Creazione/revisione/aggiornamento di procedure/template specifiche per SaMD
- UDI/ GUDID
- Registrazione del prodotto (registrazione del software)
- Registrazione dello stabilimento
- Elenco dei dispositivi
- HA Risposta alle interrogazioni - Servizi SaMD
Perché Freyr?
Domande frequenti (FAQ)
La regolamentazione del software medicale è supervisionata da diversi enti normativi a livello mondiale, tra cui la FDA negli Stati Uniti, l'EMA in Europa e la PMDA in Giappone. Queste agenzie classificano il software medico in base al rischio e stabiliscono linee guida per la sicurezza, la qualità e l'efficacia. È richiesta la conformità agli standard ISO, quali ISO 13485 e 62304.
La determinazione della classificazione del rischio del Software as a Medical Device SaMD) implica la valutazione di fattori quali l'uso previsto e il danno potenziale. I SaMD sono classificati come i dispositivi medici tradizionali in base all'importanza delle informazioni fornite alle decisioni sanitarie e allo stato della situazione o della condizione sanitaria come non grave, grave e critica. Le linee guida normative e la consultazione di esperti sono fondamentali in questo processo, per garantire la conformità e la sicurezza del paziente.
Il termine SaMD indica un software destinato a essere utilizzato per uno o più scopi medici, senza essere parte di un dispositivo medico fisico. Funziona su piattaforme informatiche generiche come smartphone, tablet o personal computer. D'altra parte, SiMD è un software che è un componente integrale di un dispositivo medico fisico, contribuendo alla sua funzionalità e alle sue prestazioni. Il SiMD non può essere utilizzato in modo indipendente e si affida al dispositivo medico associato per svolgere la sua funzione.
Un software che è incorporato come parte di un dispositivo medico hardware ed è necessario per gestire lo scopo medico previsto NON è considerato un SaMD.
La tempistica per raggiungere la conformità alla SaMD è influenzata dalla classe di rischio e dai requisiti normativi. Tuttavia, con la giusta assistenza normativa è possibile garantire un processo di conformità più agevole con rischi minimi.
Registrazione dei dispositivi medici
- Strategia regolatoria globale per le SaMD.
- Supporto normativo e di market intelligence.
- Servizi di classificazione e registrazione dei prodotti per i SaMD.
- Supporto normativo per i documenti di sviluppo dei prodotti SaMD .
- Servizi di consulenza sugli studi di valutazione clinica SaMD .
- Gestione delle modifiche post-approvazione.
- Servizio di rappresentanza locale.

- Strategia regolatoria globale per le SaMD.
- Supporto normativo e di market intelligence.
- Servizi di classificazione e registrazione dei prodotti per i SaMD.
- Supporto normativo per i documenti di sviluppo dei prodotti SaMD .
- Servizi di consulenza sugli studi di valutazione clinica SaMD .
- Gestione delle modifiche post-approvazione.
- Servizio di rappresentanza locale.





