![]()
Panoramica sulla registrazione dei dispositivi medici nel Regno Unito
Dopo la Brexit, il Regno Unito sta ancora modificando e aggiungendo i propri regolamenti per i dispositivi medici. I regolamenti da seguire nel paese sono suddivisi geograficamente – Gran Bretagna (GB) e Irlanda del Nord (NI). Medicines and Healthcare Products Regulatory Agency (MHRA) è l'autorità di regolamentazione che si occupa dei dispositivi medici. L'Irlanda del Nord è tenuta a conformarsi al Regolamento sui Dispositivi Medici dell'Unione Europea (EU MDR) 2017/745 e al Regolamento sui Dispositivi Diagnostici In Vitro (IVDR) 2017/746. I produttori non britannici devono nominare una Persona Responsabile del Regno Unito (UK RP) per aiutarli a conformarsi a questi regolamenti e garantire un processo di registrazione dei dispositivi medici nel Regno Unito di successo.
![]()
Autorità di regolamentazione: Agenzia di regolamentazione dei medicinali e dei prodotti sanitariMHRA)![]()
Regolamento: Regolamenti sui dispositivi medici (MDR) 2002*![]()
Percorso normativo: Marcatura CE seguita da notifica![]()
Rappresentante autorizzato: Persona Responsabile del REGNO UNITO (UK RP) per i produttori non del REGNO UNITO![]()
Requisito del SGQ: ISO 13485:2016![]()
Valutazione dei dati tecnici: Organismi approvati dal Regno Unito per la marcatura UKCA![]()
Marcature valide: GB - UKCA o CE & NI - CE o CE + UKNI![]()
Formato di presentazione: Carta![]()
Formato di presentazione: In linea![]()
*Il futuro regolamento per i dispositivi medici si applicherà a partire dal 1° luglio 2025.
La classificazione dei dispositivi medici nel Regno Unito
Le classificazioni dei dispositivi medici del Regno Unito si basano sulla UK MDR 2002. La classificazione del dispositivo è la prima fase dell'intero processo di approvazione e lancio sul mercato.
Classificazione dei dispositivi medici
| Classe | Il rischio |
|---|---|
| Classe I | Basso |
| Classe IIa | Medio |
| Classe IIb | Medio |
| Classe III | Alto |
Classificazione IVD
- IVD generale
- Autodiagnosi degli IVD
- IVD considerati nell'elenco A dell'allegato II
- IVD considerati nell'elenco B dell'allegato II
La nostra azienda è specializzata nella classificazione dei dispositivi medici. Finora Freyr ha completato con successo la classificazione dei dispositivi per oltre 50 aziende nel Regno Unito.
Servizi della persona responsabile del Regno Unito (UKRP)
I produttori non-UK sono ora tenuti a nominare obbligatoriamente un UKRP per immettere i prodotti sul mercato.
Freyr si è registrato con successo e può ora agire come UKRP. Per maggiori dettagli sui nostri servizi UKRP, visitate il sito www.ukrpservices.com.
Registrazione dei dispositivi medici
I produttori dovranno ora nominare un organismo approvato dal Regno Unito per ottenere la marcatura UKCA. Sebbene la marcatura CE sia consentita, è applicabile solo per un certo periodo di tempo. Le tempistiche di transizione sono indicate di seguito –
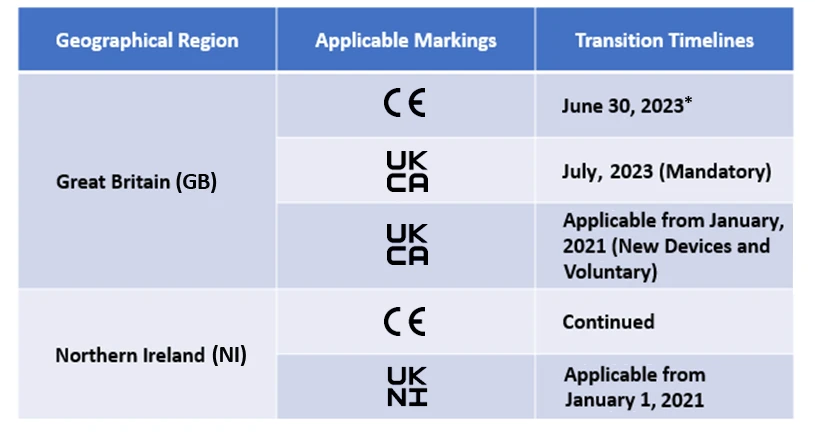
Freyr sta attualmente supportando molti produttori nelle transizioni post-Brexit.
*L'attuale calendario di transizione per la marcatura CE ai sensi dell'EU MDR IVDR è riconosciuto dall'UK MHRA e le tempistiche varieranno a seconda dell'ambito di applicazione dei dispositivi.
Requisiti per la sorveglianza post-vendita
I requisiti di sorveglianza post-commercializzazione previsti dall'MDR 2002 del Regno Unito sono piuttosto stringenti per garantire la sicurezza e l'efficacia per il paziente/utente. Attualmente, le attività di PMS comprendono la segnalazione di incidenti/riscontri all'MHRA. L'MHRA ha pubblicato una guida completa al riguardo.
Registrazione dei dispositivi medici nel Regno Unito
Competenza di Freyr
- Classificazione dei dispositivi medici nel Regno Unito
- Supporto alla transizione normativa per il post-brexit
- Supporto normativo per la notifica dell'MHRA del Regno Unito
- Persona responsabile del Regno Unito (UKRP)
- Agenzia sanitaria e Organismo approvato: collegamento e supporto