Registrazione dei dispositivi medici negli EAU Panoramica
Gli Emirati Arabi Uniti (EAU), un importante Paese membro del CCG, dispongono di un sistema sanitario avanzato. Il suo potenziale di mercato è comprovato e in costante crescita, governato dal Dipartimento di Controllo dei Farmaci del Ministero della Salute e della Prevenzione (MOHAP). La governance centralizzata e le barriere linguistiche sono i principali ostacoli alla registrazione dei dispositivi medici negli EAU, insieme alle complessità linguistiche e alla mancanza di canali di comunicazione efficienti con le autorità sanitarie.
![]()
Autorità di regolamentazione: Dipartimento per il controllo dei farmaci del Ministero della Salute e della Prevenzione (MOHAP)![]()
Regolamento: Linee guida per la registrazione dei dispositivi medici negli EAU![]()
Percorso normativo: Registrazione del prodotto![]()
Rappresentante autorizzato: È necessario un rappresentante autorizzato locale degli Emirati Arabi Uniti![]()
Requisito del SGQ: ISO 13485:2016![]()
Valutazione dei dati tecnici: Comitato per la registrazione dei dispositivi medici![]()
Validità della licenza: 5 anni![]()
Requisiti di etichettatura: Allegato 2 (2.5) delle linee guida per la registrazione dei dispositivi medici degli Emirati Arabi Uniti.![]()
Formato di presentazione: Carta![]()
Lingua: Inglese
Classificazione dei dispositivi medici degli EAU
Gli EAU hanno regole di classificazione separate per i dispositivi medici e gli IVD. Le norme di classificazione dei dispositivi medici degli Emirati Arabi Uniti sono in linea con le norme di classificazione delle direttive sui dispositivi medici dell'UE. Le classi di dispositivi secondo le regole di classificazione degli EAU sono le seguenti
| Criteri di rischio | Classe del dispositivo medico |
|---|---|
| Basso rischio | I |
| Rischio basso e moderato | IIa |
| Rischio moderato-alto | IIb |
| Alto rischio | III |
| Criteri di rischio | Classe IVD |
|---|---|
| Basso rischio individuale e basso rischio per la salute pubblica | A |
Rischio individuale moderato e/o basso rischio per la salute pubblica | B |
Rischio individuale elevato e/o Rischio moderato per la salute pubblica | C |
| Alto rischio individuale e alto rischio per la salute pubblica | D |
Rappresentante autorizzato locale degli EAU
I produttori stranieri, senza un ufficio fisico, devono nominare un Rappresentante Locale (LR) per agire per loro conto. Il rappresentante locale dovrebbe essere autorizzato dal Ministero della Salute come negozio medico o ufficio scientifico (nel caso di ufficio scientifico, le attività di importazione e distribuzione dovrebbero essere eseguite da un negozio medico autorizzato nominato). I richiedenti possono nominare il loro distributore come Rappresentante Locale. Tuttavia, avere un Rappresentante Locale indipendente, senza interessi commerciali, fornirebbe la flessibilità necessaria per nominare più distributori negli Emirati Arabi Uniti. I dettagli sia del LR che del distributore devono essere forniti durante la registrazione del dispositivo.
Processo di classificazione ufficiale con il MoHAP degli EAU
Il MoHAP degli Emirati Arabi Uniti ha introdotto un servizio di classificazione ufficiale, particolarmente utile quando non si è sicuri che il proprio prodotto debba essere registrato. Questo servizio classifica i prodotti di ogni tipo e forma in base alla loro presentazione, composizione, uso e design. I requisiti possono variare a seconda della natura del prodotto, della classe di rischio e dello status normativo.
La lettera di classificazione indica se un prodotto deve essere registrato presso il MOHAP. Se è richiesta la registrazione, il prodotto deve essere registrato in base alla classe identificata nella lettera di classificazione. Questa lettera è valida per tre anni dalla data di emissione.
I risultati della classificazione ufficiale possono essere:
- Non richiede la registrazione al MOHAP
- Autorizzato dal MOHAP degli Emirati Arabi Uniti come dispositivo medico, limitato all'uso professionale.
- Autorizzato dal MOHAP degli Emirati Arabi Uniti come dispositivo medico da banco.
Registrazione dei dispositivi medici negli EAU
Alcuni dispositivi che non richiedono la registrazione del prodotto o l'inserimento nell'elenco o l'approvazione preventiva per l'importazione. Tali prodotti esenti da registrazione o elencazione devono richiedere e ottenere un permesso di importazione per essere commercializzati negli EAU.
Per gli altri dispositivi, le importazioni non saranno sdoganate a meno che non sia stata rilasciata un'approvazione preventiva all'importazione della partita da parte del DRCD. Tali dispositivi devono essere elencati o registrati per l'importazione negli EAU.
Elenco dei dispositivi: In genere, i prodotti utilizzati negli ospedali sotto la supervisione professionale e i dispositivi di Classe I non sono sottoposti a una valutazione dettagliata e devono essere inseriti nell'elenco. L'agenzia rilascia un certificato di inclusione nell'elenco. Dopo l'inserimento nell'elenco, i dispositivi devono ottenere l'autorizzazione all'importazione per poter essere commercializzati negli EAU.
Registrazione dei dispositivi: L'attività di registrazione comprende la registrazione del sito e del prodotto.
- Registrazione del sito:il sito di produzione deve essere registrato se il dispositivo prodotto in quel sito viene importato negli EAU per la prima volta. Per i dispositivi successivi fabbricati nello stesso sito, è sufficiente la registrazione del dispositivo e non è richiesta la registrazione del sito.
- Registrazione del dispositivo:questi dispositivi sono soggetti a revisione da parte del comitato tecnico e, una volta approvati, riceveranno un certificato di licenza.
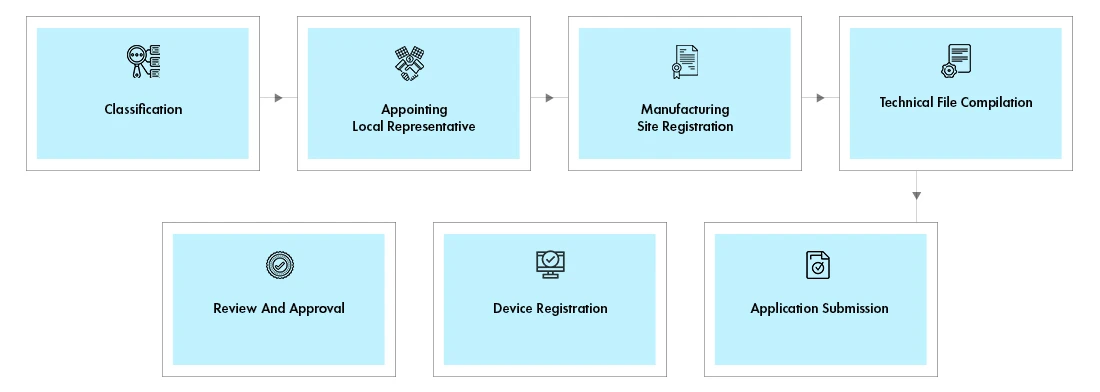
Flusso di processo
Gestione del ciclo di vita del dispositivo dopo l'approvazione
- Gestione delle modifiche post-approvazione - modifiche alle approvazioni dei dispositivi medici esistenti, come l'aggiunta di nuove varianti e accessori, l'aggiunta di nuove indicazioni d'uso, ecc.
- Mantenimento delle approvazioni e della registrazione attraverso il pagamento puntuale delle tasse amministrative e di registrazione.
- Rinnovo delle licenze
- Collegamento tra il Ministero della Salute e il produttore
- Gestione delle importazioni
Con un centro di consegna esclusivo a Dubai, Freyr una posizione autorevole nel mercato dei dispositivi medici degli Emirati Arabi Uniti e delinea la classificazione dei dispositivi oltre a decodificare le normative guida per una migliore conformità. Supportiamo i clienti nella compilazione dei documenti secondo gli standard e garantiamo così approvazioni rapide. Freyr una gamma completa di servizi normativi relativi alla commercializzazione di dispositivi di successo.
Sintesi
| Tipo di dispositivo | Elenco dei dispositivi | Registrazione del dispositivo | Licenza di importazione |
|---|---|---|---|
Dispositivo esonerato dall'approvazione pre-importazione (Come elencato nell'Allegato 3) | NA | NA | SÌ |
| SÌ | NA | SÌ |
| Tutti gli altri dispositivi | NA | SÌ | SÌ |
Competenza di Freyr
- Intelligenza normativa
- Due diligence normativa
- Classificazione formale dei dispositivi medici
- Registrazione del dispositivo
- Rappresentanza autorizzata dagli EAU
- Supporto alla traduzione
- Supporto per l'etichettatura
- Identificazione e qualificazione dei distributori
- Gestione delle modifiche successive all'approvazione
- Rinnovo e trasferimento della licenza
- Sdoganamento