

Panoramica sulla registrazione dei dispositivi medici in Turchia
Il mercato turco dei dispositivi medici ha registrato una crescita significativa e costante nell'ultimo decennio. A partire dal 2021, la registrazione dei dispositivi medici in Turchia dovrà essere conforme al Regolamento sui dispositivi medici dell'UE (MDR) 2017/745 e al Regolamento sui dispositivi medici per la diagnostica in vitro (IVDR) 2017/746. Ciò ha favorito il commercio internazionale, inducendo diverse aziende globali a lanciare i propri dispositivi medici nel Paese.
![]()
Autorità di regolamentazione: Agenzia turca per i medicinali e i dispositivi medici (TITCK)![]()
Regolamento: Regolamenti sui dispositivi medici (MDR) 2017/745, regolamenti sui dispositivi diagnostici in vitro 2017/746![]()
Percorso normativo: La marcatura CE è obbligatoria, seguita dalla registrazione/notifica nel sistema di tracciabilità dei prodotti (UTS).![]()
Turchia Rappresentante locale autorizzato: Rappresentante Autorizzato Europeo (EAR) per produttori stranieri (non-UE/non-Turchia)![]()
Requisito del SGQ: ISO 13485:2016![]()
Valutazione dei dati tecnici: Organismo notificato per la marcatura CE![]()
Validità della licenza: Illimitato![]()
Formato di presentazione: Carta![]()
Traduzione: Documenti tradotti in turco
Classificazione del dispositivo
La Turchia segue la stessa classificazione dei dispositivi medici indicata nella EU MDR e nella IVDR EU MDR . Determinare la classificazione dei dispositivi può essere un'impresa ardua e quindi il supporto di un consulente normativo esperto è fondamentale.
Classi di dispositivi medici -
| Classe | Il rischio |
|---|---|
| Classe I | Basso |
| Classe IIa | Moderato |
| Classe IIb | Da moderato a elevato |
| Classe III | Alto |
Classi di dispositivi diagnostici in vitro
| Classe | Il rischio |
|---|---|
| Classe A | Basso |
| Classe B | Moderato |
| Classe C | Da moderato a elevato |
| Classe D | Alto |
Turchia Rappresentante locale autorizzato
Ora, a causa dell'accordo di unione doganale, i produttori dell'UE non devono nominare un rappresentante autorizzato locale per immettere i loro dispositivi sul mercato.
Altri produttori stranieri sono tenuti a nominare un Rappresentante Autorizzato Europeo (EAR) per immettere i dispositivi nel mercato turco.
Registrazione dei dispositivi medici
La marcatura CE è una conformità richiesta ai produttori per immettere il loro dispositivo nel mercato turco. La marcatura CE viene rilasciata tramite una valutazione di conformità effettuata dall'organismo notificato. Ora, la Turchia è autorizzata a nominare organismi notificati in conformità con l'EU MDR e l'IVDR.
Le aziende sono tenute a registrarsi nel sistema di registrazione centrale (MERSIS) e a registrare il dispositivo nel sistema di tracciamento dei prodotti (UTS).
Flusso di processo
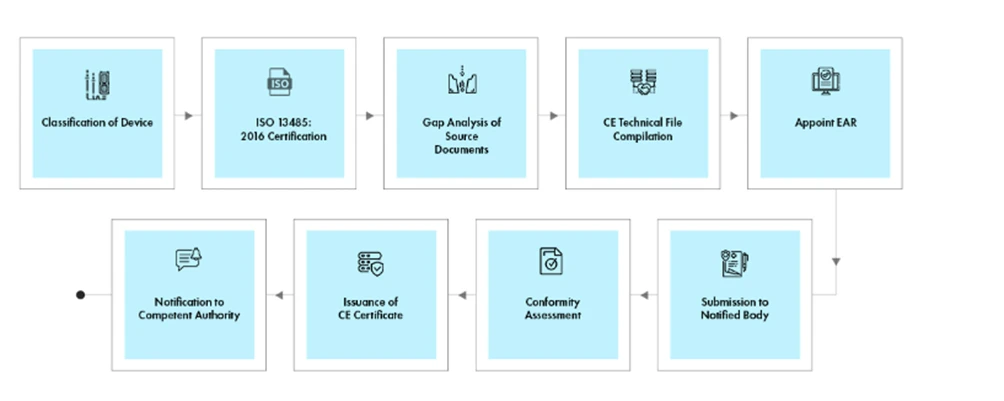
Gestione del ciclo di vita del dispositivo post-approvazione
Freyr supporta i produttori esteri nella gestione End-to-End del ciclo di vita dei dispositivi medici, incluse le attività post-approvazione, come:
- Gestione delle modifiche successive all'approvazione - modifiche alle approvazioni dei dispositivi medici esistenti, come l'aggiunta di nuove varianti, accessori, l'aggiunta di nuove indicazioni d'uso, ecc.
- Mantenimento della certificazione ISO 13485:2016 e CE
- Rinnovo delle licenze
- Collegamento tra l'organismo notificato e il produttore
Con vari organismi di autorizzazione coinvolti, i produttori esteri devono conformarsi a molteplici insiemi di regolamenti in ogni singolo processo per le approvazioni dei dispositivi. Ottenere la marcatura CE e aderire ulteriormente alle normative specifiche per stato richiede un'ampia conoscenza normativa. A volte, senza un partner normativo comprovato, orientarsi tra tutti i requisiti dei dispositivi può essere impegnativo per i nuovi operatori del mercato. Per assistere i produttori, Freyr fornisce servizi normativi End-to-End per accelerare le approvazioni per i dispositivi medici.
Competenza di Freyr
- Classificazione europea dei dispositivi medici
- Supporto del Rappresentante Autorizzato Europeo (EAR)
- Registrazione dei dispositivi e notifica dei prodotti in Turchia
- ISO 14971:2019 Consultazione sulla gestione del rischio
- Conformità ISO 13485:2016
- Revisione, compilazione e presentazione del fascicolo tecnico/di progetto CE
- Supporto alla transizione EU MDR
- Supporto alla transizione IVDR dell'UE
- Clinical Evaluation Reports (CER) per i dispositivi medici
- Relazioni di valutazione delle prestazioni (PER) per i dispositivi diagnostici in vitro
- Notifica/registrazione di dispositivi medici tramite il sistema di registrazione online
- Rapporto sulla strategia di regolamentazione dei dispositivi medici
- Supporto ai test: biocompatibilità, sicurezza elettrica, meccanica e prestazioni.
- Supporto per la conformità dell'etichettatura
- Supporto GMP
- Supporto per la sorveglianza post-vendita
