Clinical evaluation report (CER) per dispositivi medici Panoramica
Qualsiasi dispositivo destinato a essere commercializzato nell'Unione Europea (UE) deve recare un marchio CE. In conformità con l'EU MDR 2017/745 EU MDR , i requisiti per un Clinical Evaluation Report (CER), compresi i requisiti di processo e di dati, variano in base alla classe di un dispositivo e sono necessari per ottenere la certificazione CE dei dispositivi medici. I dispositivi di classe I a basso rischio possono effettuare l'autocertificazione CE. Al contrario, le altre classi di dispositivi (IIa, IIb, III) devono elaborare la certificazione del marchio CE attraverso un Organismo Notificato (NB) accreditato. Il fabbricante deve presentare il fascicolo tecnico CE all'NB per la valutazione e il rilascio dell'approvazione del marchio CE e del certificato CE. Il Clinical Evaluation Report (CER) per i dispositivi medici deve essere presentato insieme al fascicolo tecnico CE per soddisfare i requisiti della marcatura CE.
Il Clinical Evaluation Report (CER) per i dispositivi medici è uno dei rapporti che devono essere presentati insieme al fascicolo tecnico CE per soddisfare i requisiti CER.
Che cos'è un Clinical Evaluation Report (CER)?
La stesura del rapporto di valutazione clinica comprende la valutazione e l'analisi dei dati clinici relativi a un dispositivo medico per verificarne la sicurezza clinica e le prestazioni. La valutazione clinica dei dispositivi medici si basa sull'analisi completa dei dati clinici pre- e post-vendita relativi all'uso previsto. Il rapporto di valutazione clinica comprende i dati specifici del dispositivo, nonché i dati relativi ai dispositivi dichiarati equivalenti dal produttore.
Un rapporto di valutazione clinica è costituito da letteratura scientifica e dati clinici analizzati, raccolti da un'indagine clinica sul vostro dispositivo o dai risultati di altri studi su dispositivi sostanzialmente equivalenti. Il CER di un dispositivo medico dimostra che il dispositivo raggiunge lo scopo previsto senza esporre gli utenti e i pazienti a ulteriori rischi.
Il CER EU MDR deve essere aggiornato ogni anno. Nel caso in cui il dispositivo sia stato commercializzato per un periodo significativo e la sua sicurezza sia stata dimostrata senza rischi significativi, il CER può essere aggiornato ogni 2-5 anni. Qualsiasi modifica apportata al design del dispositivo e qualsiasi nuova informazione derivante dai dati PMS possono determinare un aggiornamento del rapporto CER.
La valutazione clinica dei dispositivi medici, così come inquadrata nel Clinical Evaluation Report (CER), si basa sui fattori elencati di seguito.
- Letteratura scientifica attualmente disponibile; e/o
- Indagini cliniche effettuate; oppure
- Se la dimostrazione della conformità ai requisiti essenziali basata sui dati clinici non è ritenuta appropriata.
Fasi della stesura di Clinical Evaluation Report (CER)
In riferimento al nuovo Regolamento UE sui dispositivi medici (MDR) - 2017/745, ci sono quattro (04) fasi diverse per eseguire una valutazione clinica dei dispositivi medici e preparare un Clinical Evaluation Report (CER) MDR completo.
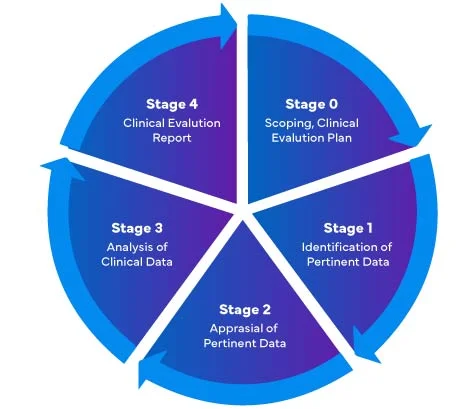
I produttori di dispositivi medici che entrano per la prima volta nel mercato dell'UE devono assicurarsi che il loro Rapporto di Valutazione Clinica sia conforme ai regolamenti EU MDR.
Freyr offre servizi End-to-End per la certificazione CE ai produttori di dispositivi, inclusa la redazione del Rapporto di Valutazione Clinica in linea con i regolamenti EU MDR 2017/745 di nuova attuazione. Con una forte esperienza regionale sui dispositivi medici dell'UE, Freyr soddisfa i requisiti specifici delle agenzie e personalizza di conseguenza il Rapporto di Valutazione Clinica.
Ottenere la consulenza di esperti per la presentazione del CER
Clinical evaluation report (CER)
- Supporto End-to-End alla redazione di Rapporti di Valutazione Clinica, inclusa la ricerca bibliografica, secondo la revisione 4 del MEDDEV 2.7/1 e le linee guida del Regolamento UE sui Dispositivi Medici (MDR).
- Elaborazione di un piano di valutazione clinica per la vostra organizzazione.
- Identificare, cercare, analizzare e mettere insieme la letteratura scientifica appropriata applicabile.
- Sviluppare un modello di rapporto di valutazione clinica per la propria organizzazione.
- Analisi delle lacune per il rapporto di valutazione clinica esistente.
- Strumento DMS che consente al vostro team di contribuire collettivamente alla stesura dei rapporti di valutazione clinica.
- Integrazione dei dati PMS.
- Sviluppare una procedura operativa standard che consenta al team di compilare i dati PMS per aggiornare i rapporti di valutazione clinica.
- Gestire gli aggiornamenti periodici dei rapporti di valutazione clinica esistenti, secondo le linee guida MDR dell'UE.
- Supporto dei dati PMS per i dispositivi esistenti sul mercato.
- Conformità alla marcatura CE e servizi di marcatura CE.

- Garantire la conformità alle recenti normative applicabili.
- Team di esperti clinici qualificati.
- Contributi interfunzionali da parte di esperti di dispositivi medici per soddisfare i requisiti.
- Servizio completo di conformità, revisione e pianificazione.





