Panoramica dei servizi di conformità IVDR UE
Il Regolamento Europeo sui Dispositivi Medico-Diagnostici in Vitro (EU IVDR) è una nuova base normativa per l'immissione di IVD sul mercato europeo. Il Regolamento EU IVDR è destinato a sostituire l'attuale Direttiva UE sui dispositivi medico-diagnostici in vitro (UE 98/79/CE). In quanto regolamento europeo, sarà efficace in tutti i Member States dell'UE e negli stati dell'Associazione Europea di Libero Scambio (EFTA) senza la necessità di essere recepito nella legislazione dei rispettivi Stati. In qualità di partner normativo comprovato, Freyr fornisce servizi di conformità EU IVDR e servizi di consulenza IVDR ai produttori per i Regolamenti sui Dispositivi Medico-Diagnostici in Vitro conformi e la marcatura CE.
Il regolamento IVDR - Cronologia della transizione
Il nuovo IVDR dell'UE, che entrerà in vigore il 26 maggio 2022, è considerato un cambiamento significativo per la sorveglianza regolamentare degli IVD. Di seguito una panoramica della transizione dell'IVDR.
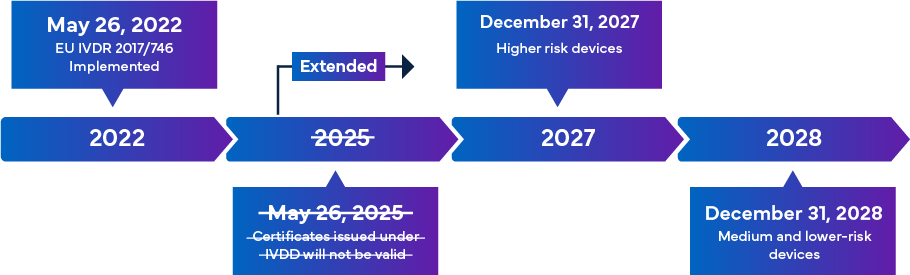
Una volta pienamente attuate, le norme di conformità IVDR garantiscono che quasi tutti i dispositivi medico-diagnostici in vitro (IVD) che entrano nel mercato europeo siano soggetti alla revisione dell'organismo notificato (NB) IVDR e alla certificazione di marcatura CE come parte del processo di approvazione degli IVD. In questo scenario, oltre ad allinearsi alla classificazione IVDR aggiornata, vi è l'esigenza immediata per i produttori di rivedere efficacemente la documentazione tecnica chiave per una registrazione e marcatura CE degli IVD di successo. I requisiti IVDR variano in base alla classe di rischio dell'IVD. In generale, per la certificazione degli IVD, i produttori devono eseguire:
- Revisione dei fascicoli tecnici secondo il regolamento IVDR (Regolamento UE 2017/746).
- Preparazione della relazione di valutazione delle prestazioni (PER) per tutti gli IVD.
- Follow-up delle prestazioni post-commercializzazione (PMPF) come previsto dall'Allegato XIII, Parte B, della IVDR.
- Segnalazione di vigilanza ai sensi dell'art. 82 dell'IVDR
Freyr supporta i clienti nell'esecuzione di una revisione sistematica della letteratura scientifica e nell'assistenza alla pianificazione e preparazione di un PER per la conformità al Regolamento sui Dispositivi Medico-Diagnostici in Vitro. Freyr dispone di esperti dedicati che forniscono servizi di consulenza IVDR e supporto per la Post-market Surveillance (PMS), che è parte integrante del sistema di gestione della qualità del produttore insieme al Post-market Performance Follow-up (PMPF).
Ottenete una consulenza esperta sulla vostra conformità agli standard IVDR dell'UE
Servizi di conformità IVDR UE Competenza e vantaggi
- Piano di transizione per la conformità IVDR
- Revisione tecnica e analisi delle lacune dei requisiti IVDR per i GSPR (General Safety and Performance Requirements).
- Supportare la compilazione del fascicolo tecnico secondo i requisiti IVDR
- Rapporti di validità scientifica basati sulla letteratura e/o su dati interni all'azienda.
- Rapporti sulle prestazioni cliniche basati sulla letteratura e/o sui dati interni all'azienda.
- Evidenze cliniche o rapporti di valutazione delle prestazioni
- Protocolli e rapporti di follow-up delle prestazioni post-vendita (PMPF)
- Protocolli e rapporti di Post-Market Surveillance (PMS)
- Scrivere/revisionare altri documenti come foglietto illustrativo/IFU (Istruzioni per l'uso), Istruzioni di riferimento rapido (QRI), Manuale d'uso/di funzionamento ecc.

- Conformità IVDR garantita, registrazione IVD e marchio CE
- Forte comprensione della normativa e competenza nelle aree di impatto chiave dell'IVDR dell'UE
- Un modello di consegna fortemente orientato alla gestione dei progetti per garantire il rispetto delle scadenze.
- Esperti NB interni (revisione del rapporto da parte dei revisori interattivi NB)
- Team focalizzati con competenze trasversali su specifiche aree di impatto e categorie di dispositivi
- Contributi interfunzionali da parte di esperti di dispositivi medici per la conformità ai requisiti
- Servizi completi di conformità, revisione e pianificazione
- Forte esperienza nel mantenere la coerenza dei risultati (tempo e qualità).