
Panoramica sulla registrazione dei dispositivi medici in Nuova Zelanda
I dispositivi medici in Nuova Zelanda sono regolamentati dall'Autorità neozelandese per la sicurezza dei medicinali e dei dispositivi medici (Medsafe) ai sensi del Medicines Regulations 1984, del Medicines Act 1981 e del Medicines (Database of Medical Devices) Regulations 2003. Sebbene non sia necessaria l'approvazione prima dell'immissione in commercio, è necessario inserire i prodotti nella banca dati elettronica WAND (Web Assisted Notification of Devices) entro 30 giorni dal lancio commerciale. Medsafe può richiedere la documentazione comprovante la sicurezza e l'efficacia, come la certificazione da parte di enti riconosciuti come un organismo notificato dell'UE o Health Canada.
Il team di esperti di regolamentazione dei dispositivi medici di Freyr vanta una notevole esperienza nel guidare le aziende di dispositivi medici attraverso il processo di registrazione Medsafe per i dispositivi medici in Nuova Zelanda.
![]()
Autorità di regolamentazione: Autorità per la sicurezza dei dispositivi medici (Medsafe)![]()
Regolamento:Regolamento sui medicinali (banca dati dei dispositivi medici), 2003
Legge sui medicinali 1981
Regolamento sui medicinali 1984![]()
Percorso normativo: Sistema elettronico di notifica via web dei dispositivi (WAND)![]()
Rappresentante autorizzato: Dispositivo medico Sponsor![]()
Requisito del SGQ: Certificazione ISO 13485:2016![]()
Valutazione dei dati tecnici: Autorità per la sicurezza dei dispositivi medici (Medsafe)![]()
Validità della licenza: Gli elenchi dei dispositivi in Nuova Zelanda non hanno scadenza. I dispositivi ritenuti una grave minaccia per il pubblico possono essere ritirati dal mercato.![]()
Requisiti di etichettatura: Regolamento 12(4) dei Regolamenti sui medicinali del 1984 e GHTF/SG1/N43:2005![]()
Formato di presentazione: Sistema elettronico di notifica via web dei dispositivi (WAND)![]()
Lingua: Inglese
Classificazione dei dispositivi medici in Nuova Zelanda
I dispositivi medici in Nuova Zelanda sono classificati in base al rischio nelle classi I, IIa, IIb, III e AIMD, secondo i criteri dell'International Medical Device Regulators Forum (IMDRF). Questa classificazione influisce sulla quantità di controlli normativi necessari. La classificazione si basa su caratteristiche quali la destinazione d'uso del dispositivo, la durata del contatto con il corpo, l'invasività e il fatto che sia attivo o inattivo. I dispositivi di classe superiore sono soggetti a un controllo normativo più severo. Medsafe è l'ente normativo della Nuova Zelanda che supervisiona queste classificazioni e normative.
| Classificazione dei dispositivi medici Medsafe diversi dagli IVD Classe | Il rischio |
|---|---|
| Classe I Basic | Basso rischio |
| Misurazione di Classe I | Basso rischio |
| Classe I sterile | Basso rischio |
| Classe IIa | Rischio medio-basso |
| Classe IIb | Rischio medio-alto |
| Classe III e dispositivo medico impiantabile attivo (AIMD) | Alto rischio |
| Classificazione Medsafe IVD | Il rischio |
|---|---|
| A partire da luglio 2014, Medsafe non riconosce alcun sistema di classificazione del rischio per gli IVD. Tutti gli IVD notificati a WAND devono utilizzare il codice di classificazione del rischio dell'IVD. Il Direttore Generale della Sanità ha autorizzato l'esenzione per gli IVD in base all'Allegato 1, paragrafo (i) del Regolamento 2003 sui Medicinali (Database dei Dispositivi Medici). Tuttavia, i fornitori di IVD possono notificare volontariamente i loro dispositivi alla banca dati. | |
Rappresentante autorizzato per i dispositivi medici/Sponsor
Il rappresentante autorizzato è definito Sponsor e funge da intermediario tra il produttore e Medsafe. Gli sponsor fungono da rappresentanti regolatori per i prodotti commercializzati in Nuova Zelanda, presentando le domande WAND e fungendo da punto di contatto primario tra il produttore e Medsafe per tutte le questioni relative al prodotto. Inoltre, Medsafe ritiene lo Sponsor responsabile degli sforzi di vigilanza.
Registrazione dei dispositivi medici in Nuova Zelanda
La procedura di registrazione dei dispositivi medici in Nuova Zelanda e la procedura di inserimento nell'elenco WAND in Nuova Zelanda variano a seconda della classe del dispositivo.
Dispositivi di Classe I - Per i dispositivi non sterili e non misuratori di Classe I è necessaria una dichiarazione di conformità del produttore; tuttavia, raramente viene presentata a un ente normativo. Invece, lo sponsor (o il fornitore) deve inserire i dettagli del dispositivo nel database Web Assisted Notification of Devices (WAND) come parte del processo di notifica Medsafe.
Altri dispositivi di classe
In Nuova Zelanda, gli sponsor o i fornitori hanno il compito di garantire la conformità dei dispositivi medici a standard come la ISO 13485:2016. In genere non è richiesta la presentazione diretta a Medsafe di una dichiarazione di conformità, di una certificazione QMS o di prove di produzione. Tuttavia, la conservazione di questa documentazione è fondamentale per dimostrare la conformità su richiesta.
Medsafe dà priorità alla sorveglianza post-market rispetto all'approvazione dettagliata pre-market dei dispositivi medici. Sebbene gli audit non vengano condotti di routine durante la fase di notifica, Medsafe può avviarli per i dispositivi a rischio più elevato o a seguito di attività di vigilanza e di segnalazioni di eventi avversi, garantendo sicurezza e conformità costanti.
Una volta che un dispositivo è stato notificato attraverso il database WAND, può essere commercializzato in Nuova Zelanda, a condizione che il fornitore soddisfi costantemente i regolamenti di Medsafe. Ciò richiede una conformità continua, in particolare con gli standard di monitoraggio post-market e di segnalazione degli incidenti. Gli esperti di dispositivi medici di Freyr supportano i servizi relativi alla gestione di questi requisiti normativi, assicurando che le aziende mantengano la conformità durante l'intero ciclo di vita del prodotto.
Flusso di processo
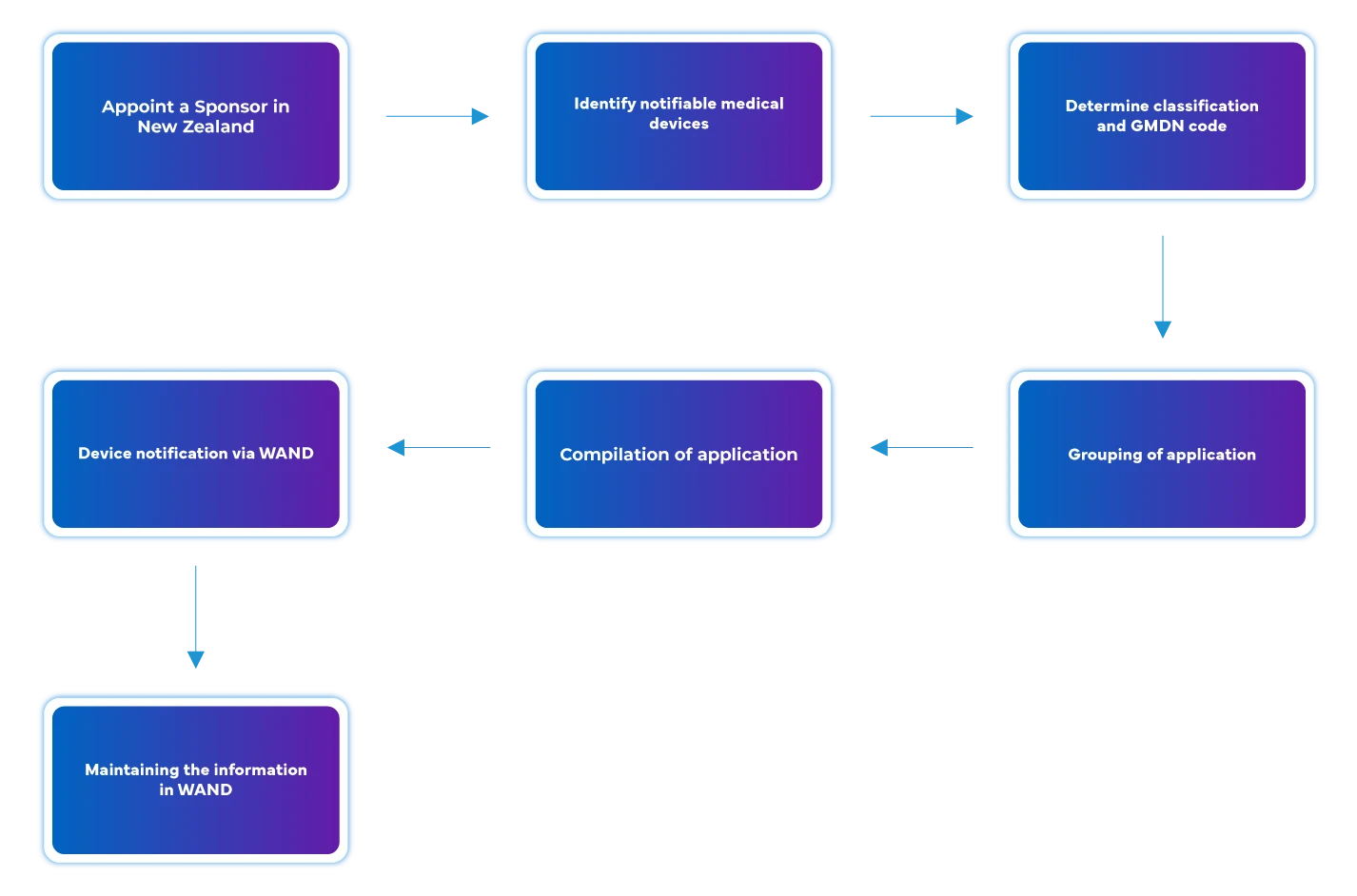
Gestione del ciclo di vita del dispositivo dopo l'approvazione
Freyr supporta i produttori esteri nella gestione End-to-End del ciclo di vita dei dispositivi medici, incluse le attività post-approvazione, notificando le autorità neozelandesi tramite WAND, come –
- Gestione delle modifiche successive all'approvazione - modifiche alle approvazioni dei dispositivi medici esistenti, come l'aggiunta di nuove varianti e accessori, l'aggiunta di nuove indicazioni d'uso, ecc.
- Mantenimento delle approvazioni e della registrazione.
Dotata di un team di professionisti normativi, Freyr offre un supporto completo ai produttori per mantenere gli standard di qualità e sicurezza richiesti per l'approvazione del mercato. Gli specialisti di intelligence normativa dell'azienda monitorano meticolosamente gli aggiornamenti delle normative, assicurando che i clienti siano ben informati sulle azioni necessarie per mantenere la conformità dei loro prodotti agli standard attuali.
Sintesi
| Il rischio | Classe del dispositivo | Audit del SGQ | Percorso normativo | Tempistiche Medsafe | Validità della registrazione (anni) |
|---|---|---|---|---|---|
| Basso rischio | Classe I Basic | Conformità alla norma ISO 13485:2016 Nota: Medsafe non richiede audit del SGQ, ma raccomanda vivamente di seguire la norma ISO 13485:2016 per la qualità e la sicurezza. Medsafe ha l'autorità di condurre audit del SGQ per qualsiasi classe di dispositivi se emergono problemi di sicurezza o di qualità. | Elenco WAND (notifica) | 1 settimana |
Nessuna data di scadenza |
| Basso rischio | Misurazione di Classe I | Elenco WAND (notifica) | |||
| Basso rischio | Classe I sterile | Elenco WAND (notifica) | |||
| Rischio medio-basso | Classe IIa | Elenco WAND (notifica) | |||
| Rischio medio-alto | Classe IIb | Elenco WAND (notifica) | |||
| Alto rischio | Classe III | Elenco WAND (notifica) |
Nota: in base alla legislazione vigente, gli elenchi dei dispositivi in Nuova Zelanda non hanno scadenza, ma i dispositivi che si ritiene rappresentino un rischio inaccettabile per il pubblico possono essere ritirati dal mercato. Tuttavia, l'attuale legislazione potrebbe essere rivista entro il 2026/2027.
Competenza di Freyr
- Supporto alla registrazione dei dispositivi medici end-to-end.
- Supporto LR
- Elenco WAND
- Supporto per l'etichettatura
- Gestione delle modifiche successive all'approvazione
- Trasferimento della licenza
- Presentazione e servizi di collegamento con WAND