Registrazione di SaMD negli US – Panoramica
Risposte in questa pagina
- Il mio software è un SaMD?
- Quale classe di rischio di SaMD richiede una 510(k)?
- Registrazione e conformità SaMD - il processo 510(k)
- Per quanto tempo è valida l'autorizzazione?
Il mio software è un SaMD?
Secondo l'International Medical Device Regulators Forum (IMDRF), il SaMD è un:
- Software destinato a essere utilizzato per uno o più scopi medici.
- Svolge queste funzioni senza essere parte di un dispositivo medico hardware.
Quale classe di rischio di SaMD richiede una 510(k)?
Determinare la classificazione SaMD per il vostro software è un passo importante nel processo di registrazione. Una volta che il vostro software è stato classificato come SaMD, diventa essenziale comprendere il percorso normativo richiesto per ottenere l'ingresso nel mercato nel US. SaMD è tipicamente categorizzato in diverse classi, in base ai loro livelli di rischio. Il SaMD di Classe II è considerato a rischio moderato, richiede un'autorizzazione 510(k) e si basa sulla dimostrazione di sostanziale equivalenza con il dispositivo precursore legalmente commercializzato. Il processo di autorizzazione garantisce che il vostro SaMD sia sostanzialmente equivalente ai dispositivi esistenti, il che aiuta a garantirne la sicurezza e l'efficacia prima che venga commercializzato.
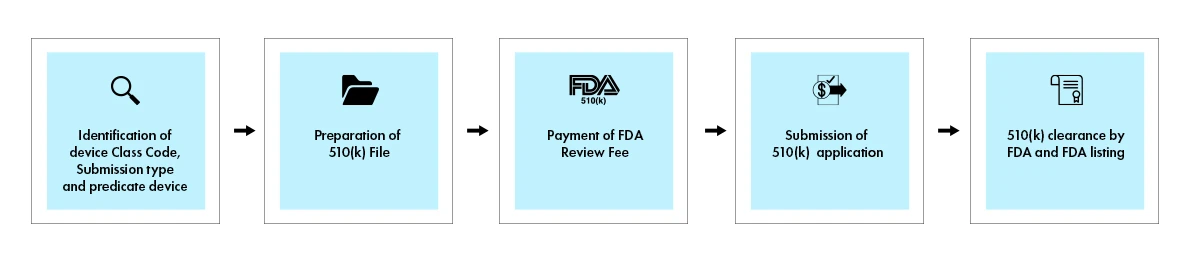
Registrazione e conformità SaMD - il processo 510(k)
Il processo 510(k) comporta una sottomissione completa che dimostra un'equivalenza sostanziale a un dispositivo precursore legalmente commercializzato. Quando viene presa una decisione, la US Food and Drug Administration (FDA) invia una lettera di decisione al richiedente via email. Una domanda 510(k) che riceve una lettera di decisione di equivalenza sostanziale è considerata "approvata". Viene quindi elencata nel database 510(k), con il riepilogo 510(k) allegato. La figura seguente fornisce una panoramica visiva dei passaggi chiave coinvolti nel processo 510(k).
Per quanto tempo è valida l'autorizzazione?
Un'autorizzazione 510(k) rimane valida fino a quando non vengono apportate modifiche significative al dispositivo o alle normative applicabili. Tuttavia, è importante notare che la US FDA può richiedere rapporti periodici o informazioni aggiuntive per garantire la conformità e la sicurezza continue.
In conclusione, la registrazione SaMD richiede una comprensione approfondita della classificazione SaMD , della conformità SaMD e dei processi normativi. Rivolgersi a un servizio di consulenza SaMD può fornire una guida esperta per navigare tra le complessità e garantire un esito positivo della registrazione.
Registrazione di SaMD negli US
- Strategia normativa US FDA completa per SaMD.
- Classificazione SaMD .
- Identificazione del dispositivo predicato.
- Stabilire l'equivalenza sostanziale con il dispositivo predicato.
- Analisi delle lacune per la conformità alla US FDA.
- Compilazione del fascicolo tecnico 510(k), secondo la Guida alle Presentazioni Premarket per il Software della FDA US.
- Creazione del modello eCopy/eSTAR.
- Convalida e invio del modello eCopy/eSTAR.
- Collegamento con i servizi per l'approvazione del dispositivo.
- Supporto alla risposta dell'ACR e alle carenze dell'AINN.
- Servizi di consulenza per risolvere le carenze.
- Registrazione dello stabilimento presso la FDA degli US.
- Elenco dei dispositivi e manutenzione del database FURLS.
- Servizi di rappresentanza legale (LR).

- Ampia esperienza con diverse registrazioni 510(k).
- Competenza nella compilazione del 510(k), secondo i requisiti di notifica pre-commercializzazione (510[k]) della US FDA.
- Supporto aggiuntivo per la gestione delle richieste 510(k).
- Presentazione puntuale dei prodotti.
- Aggiornato con i nuovi emendamenti della US FDA sui SaMD.