4 minuti di lettura
La segnalazione di dispositivi medici (MDR) è uno strumento di sorveglianza post-commercializzazione che la Food and Drug Administration (FDA) utilizza per monitorare le prestazioni dei dispositivi, rilevare potenziali problemi di sicurezza correlati ai dispositivi e contribuire alle valutazioni beneficio-rischio dei dispositivi. Lo scopo dell'MDR è rilevare e affrontare tempestivamente gli eventi avversi correlati ai dispositivi. Consente a medici, strutture sanitarie, produttori e consumatori di effettuare segnalazioni volontarie per comprendere la sicurezza e l'efficacia post-commercializzazione del dispositivo.
L'MDR è applicabile a tutte le classi di dispositivi medici, che sono fabbricati negli Stati Uniti d'America (USA) o importati negli USA. I produttori di dispositivi medici che intendono commercializzare i loro dispositivi negli USA devono essere conformi all'MDR, altrimenti ciò potrebbe comportare sanzioni finanziarie. È applicabile negli USA, inclusi eventi esteri, ovvero è applicabile ai dispositivi medici legalmente commercializzati negli Stati Uniti, sia fabbricati negli USA che in paesi esteri. Inoltre, esistono vari casi di applicabilità per un MDR, come:
- se un dispositivo è prodotto negli Stati Uniti, distribuito localmente e in altri mercati
- quando un dispositivo è prodotto negli Stati Uniti ma distribuito in altri mercati
- quando un dispositivo è prodotto nel paese straniero e fornito negli Stati Uniti e in altri mercati
- quando un dispositivo è fabbricato nel paese straniero e distribuito localmente e
- quando un dispositivo è oggetto di indagine negli Stati Uniti
MDR e flusso del processo di reporting
Il regolamento MDR contiene molti requisiti obbligatori per produttori, importatori e strutture che utilizzano dispositivi, per segnalare alla FDA determinati eventi avversi correlati ai dispositivi e problemi del prodotto. Il diagramma di flusso del processo fornito di seguito illustra il processo di segnalazione passo dopo passo.
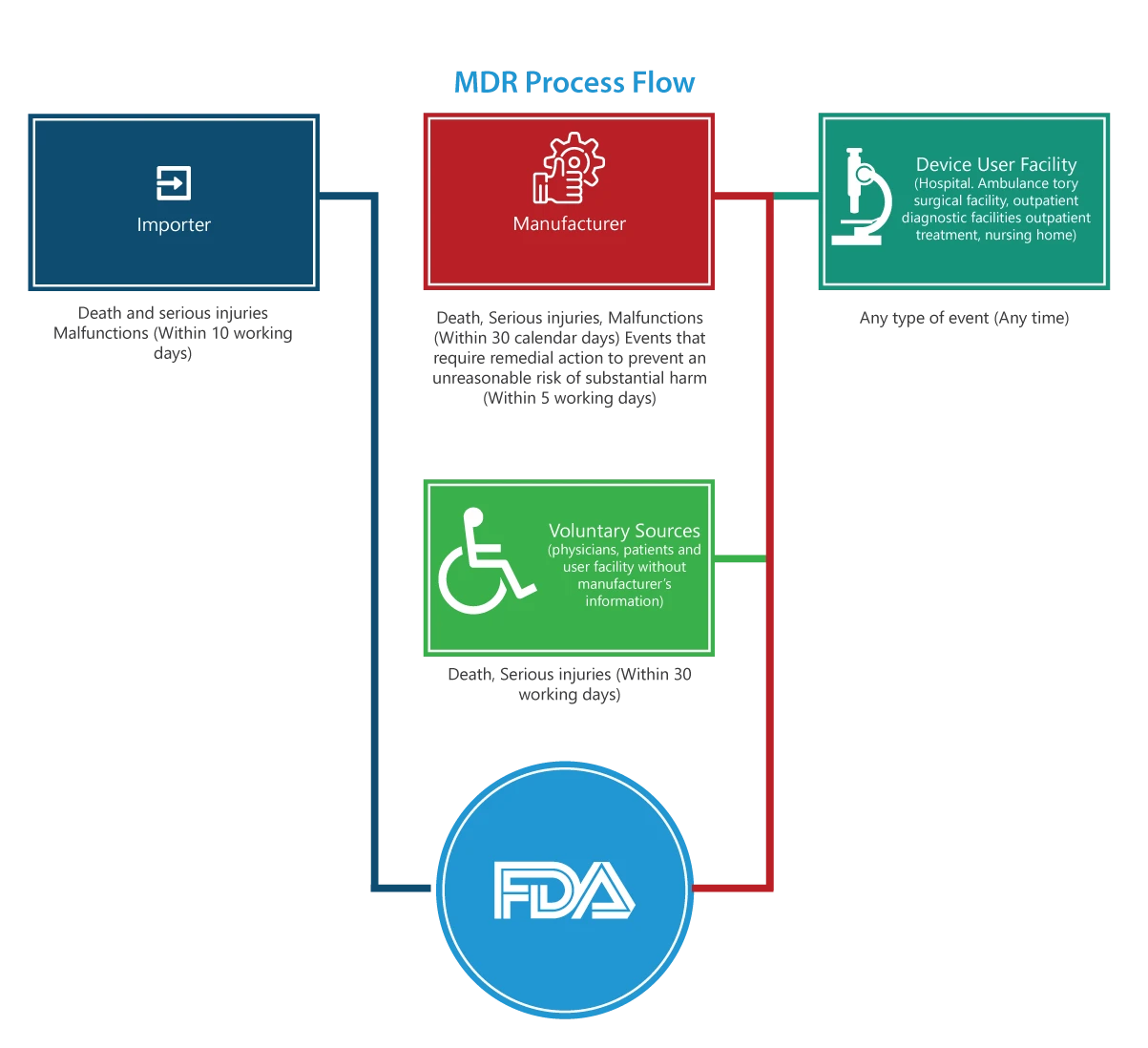
A chi si applica?
Importatori
Le segnalazioni di decessi, lesioni gravi e malfunzionamenti devono essere presentate alla FDA e al produttore entro 30 giorni lavorativi. Se il malfunzionamento può causare lesioni o decessi altrove, gli importatori devono segnalare il malfunzionamento al produttore.
produttori
Le segnalazioni di un evento (decessi, lesioni gravi e malfunzionamenti) designato dall'FDA o di un evento che richiede un'azione correttiva per prevenire un rischio irragionevole di danno sostanziale alla salute pubblica devono essere presentate all'FDA entro 5 giorni lavorativi compilando il modulo 3500A.
Struttura utilizzatrice del dispositivo (ospedale, struttura chirurgica ambulatoriale, casa di cura, struttura diagnostica ambulatoriale o struttura di trattamento ambulatoriale)
I rapporti devono essere presentati al produttore del dispositivo entro e non oltre 10 giorni lavorativi dal giorno in cui si viene a conoscenza di informazioni secondo cui un dispositivo ha o potrebbe aver causato o contribuito a una lesione grave a un paziente della struttura. Se il produttore non è noto, la struttura deve presentare il rapporto all'FDA.
Gruppi di volontariato
I pazienti, gli operatori sanitari e i consumatori che riscontrano un problema relativo a un dispositivo medico possono segnalarlo all'FDA tramite MedWatch.
eMDR
L'FDA ha imposto l'MDR elettronico (eMDR) nel 2015 per identificare le criticità relative alla qualità e all'integrità dei dati associati alla segnalazione di lesioni gravi relative a tutte le classi di dispositivi medici.
I produttori possono inviare i loro eMDR tramite un Electronic Submissions Gateway (ESG). Dal momento dell'invio, il gateway elettronico impiega fino a 48 ore per inviare una conferma. Se si verifica un errore durante l'invio del rapporto, verrà visualizzato un messaggio per apportare le correzioni.
eMDR - Quali sono i suoi benefici?
eMDR offre molteplici vantaggi rispetto al meccanismo di segnalazione manuale (ossia, MDR). Di seguito sono elencati alcuni notevoli vantaggi su cui produttori / agenzie / pazienti possono contare:
- Lo strumento di presentazione eMDR migliora la collaborazione tra un'organizzazione, l'agenzia sanitariaFDA) e i pazienti.
- L'eMDR consente di risparmiare sui costi. L'automazione riduce la necessità di spese amministrative e di comunicazione tradizionale; contribuisce ad accelerare il processo e favorisce un'efficace segnalazione degli eventi, con conseguente interazione immediata con l'FDA.
- I processi manuali comportano una notevole quantità di documenti cartacei, possono essere lunghi e difficili da tracciare ed elaborare. L'invio di eMDR è automatizzato e centralizzato. I documenti possono essere recuperati facilmente, risparmiando molto tempo durante la revisione.
- L'eMDR consente alle parti di segnalare rapidamente gli errori di presentazione, anziché ricorrere alla corrispondenza manuale con l'FDA, che richiede molto tempo.
- eMDR funge da unico punto di accesso per elaborare tutte le richieste elettroniche in un ambiente altamente protetto ed è vantaggioso perché i reclami dell'organizzazione possono essere collegati direttamente al modulo MedWatch e integrati nel gateway della FDA.
eMDR e il flusso del processo di segnalazione
Il regolamento eMDR contiene requisiti obbligatori per produttori, importatori e strutture utilizzatrici di dispositivi di segnalare alla FDA determinati eventi avversi correlati ai dispositivi e problemi del prodotto. Il diagramma di flusso seguente descrive il processo di segnalazione passo dopo passo.
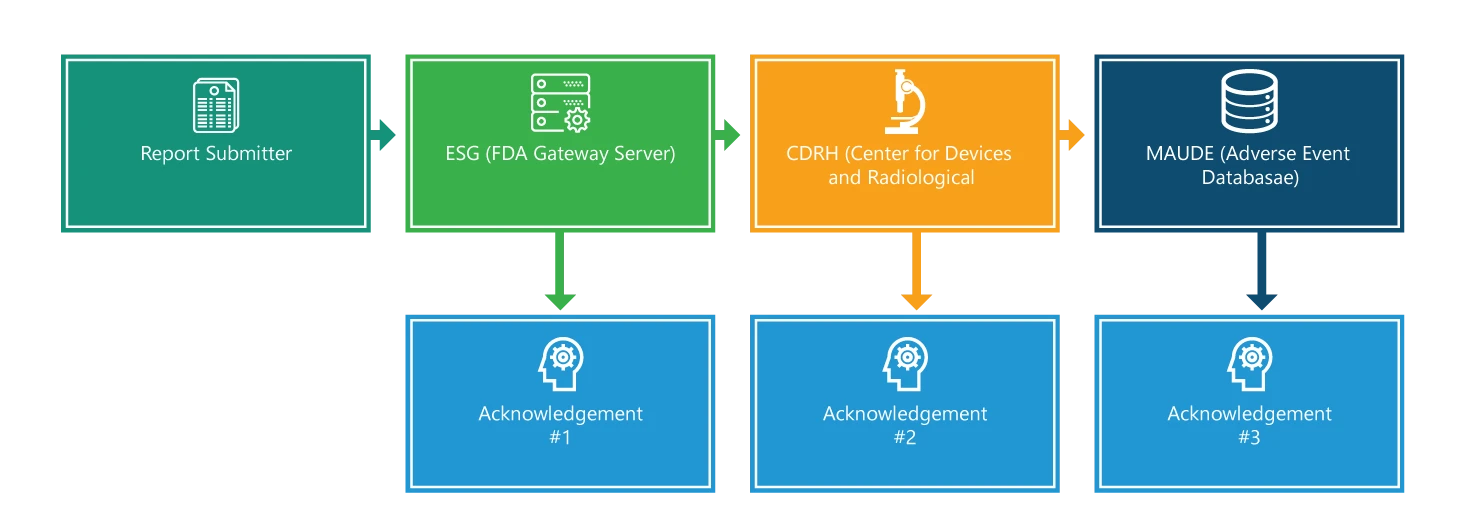
Il processo di segnalazione comprende quattro fasi. Ad eccezione della prima fase, ogni fase è riconosciuta. Inoltre, ogni fase è corredata da informazioni aggiuntive che facilitano il processo.
Fase 1: invio del rapporto
Invio di un eMDR. Per effettuare un invio, è necessario disporre di una firma elettronica e assicurarsi che i nomi dei file di invio includano solo un punto, utilizzato per indicare l'estensione del tipo di file (ad esempio 555xml o 555.pdf). Tuttavia, i tempi di consegna e di elaborazione della domanda dipendono dalle dimensioni complessive dell'invio; gli invii più grandi richiedono tempi più lunghi per essere consegnati ed elaborati.
Fase 2: Gateway di invio elettronicoESG)
Quando la vostra richiesta arriva all'ESG, dovreste ricevere rapidamente una conferma di ricezione n. 1, a meno che l'ESG non sia inattivo per manutenzione. Siete tenuti a controllare lo stato del vostro MDR sul sito web dell ESG .
Fase 3: CRDH
L'eMDR viene instradato automaticamente dall'ESG al Center for Devices and Radiological Health (CDRH). Una volta instradato, come nel passaggio 2, dovreste ricevere una conferma, ovvero il numero 2.
Fase 4: Esperienza del dispositivo del produttore e dell'utente (MAUDE)
Quando il CDRH convalida e aggiorna l'invio nel database degli eventi avversi (MAUDE), si prevede che l'autore dell'invio riceva una conferma di ricezione #3. Si noti che tutti gli errori che si verificano durante la convalida e il caricamento vengono registrati.
La segnalazione dei dispositivi medici (MDR) è un processo fondamentale che contribuisce a salvare vite umane e a proteggere i pazienti da rischi inutili. Garantisce che tutte le parti coinvolte nella cura del paziente siano responsabili e attente nell'utilizzo dei dispositivi.
L'eMDR facilita la stesura dei rapporti, ma la documentazione e il follow-up possono richiedere molte risorse. Per farlo bene la prima volta, rivolgetevi a us di sales@freyrsolutions.com.