2 minuti di lettura
L'obiettivo principaleFDAUS è quello di esaminare costantemente e colmare il divario tra i processi normativi per l'importazione e la vendita ininterrotta di dispositivi medici nuovi e di alta qualità nel US .FDA 1998 laFDA US ha pubblicato un programma denominato "Il nuovo paradigma 510(k): approcci alternativi per dimostrare la sostanziale equivalenza nelle notifiche pre-commercializzazione". Esso intende stabilire un percorso efficiente per la presentazione della domanda FDA (k) che contiene alcune modifiche alla domanda 510(k) già approvata. Questa nuova notifica 510(k) offre (03) tre tipi di presentazione, ovvero 510(k) speciale, 510(k) abbreviata e 510(k) tradizionale. Nel 2019, laFDA US ha pubblicato un documento guida speciale 510(k) che descrive un percorso opzionale per produttori apportano determinate modifiche ben definite ai loro dispositivi commercializzati legalmente.
Perché una 510(k) speciale?
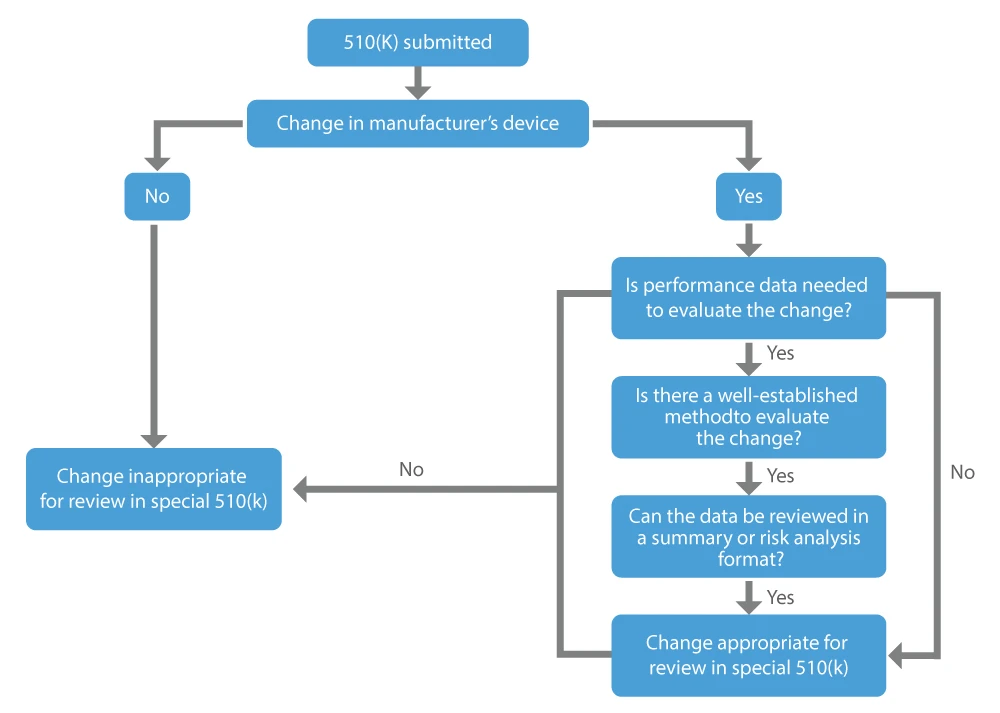
Quando un produttore desidera ottenere l'approvazione per le modifiche apportate al dispositivo già commercializzato, ovvero al dispositivo esistente, può richiedere una 510(k) speciale. I principali fattori da considerare per determinare se una modifica a un dispositivo esistente può essere appropriata per una 510(k) speciale sono i seguenti:
- La modifica riguarda il dispositivo di riferimento legalmente commercializzato dal proponente.
- Non sono richiesti dati sulle prestazioni, oppure sono disponibili metodi consolidati se si ritiene necessario valutare la modifica.
- Tutti i dati sulle prestazioni a sostegno della determinazione dell'equivalenza sostanziale possono essere esaminati in un formato di sintesi o di analisi dei rischi.
Documenti richiesti per lo speciale 510(k)
- Lettera di presentazione
- Il nome del dispositivo legalmente commercializzato (esistente) del fabbricante e il numero 510(k)
- Una descrizione dettagliata delle modifiche apportate al dispositivo che hanno comportato la presentazione di un nuovo 510(k)
- Un confronto tra il dispositivo modificato e il dispositivo autorizzato, in formato tabellare.
- Altre modifiche all'etichettatura o al design
- Una sintesi delle attività di controllo della progettazione
- Sulla base dell'analisi dei rischi, è stata identificata l'attività di verifica e/o convalida necessaria per conformarsi alla norma 21 CFR 820.30.
- Modulo per le indicazioni d'uso
- Una dichiarazione che attesti che il presentatore ha rispettato e non sta violando i requisiti della procedura di controllo della progettazione, come specificato nel 21 CFR 820.30, e che i registri sono disponibili per la revisione su richiesta.
Tempistiche di revisione speciale 510(k) da parte della FDA US
Secondo le linee guida dell'FDA"Refuse to Accept Policy for 510(k) s", la tempistica di revisione per le richieste di 510(k) speciali è di trenta (30) giorni dal ricevimento.
Quando richiedere uno speciale 510(k)?
FDA US FDA costantemente per fornire dispositivi medici sicuri ed efficaci al fine di promuovere la salute umana. Il programma speciale 510(k) è efficiente e coerente con la procedura di revisione meno onerosa che aiuta produttori stranieri produttori vendere i propri dispositivi negli Stati Uniti e consente ai pazienti di accedere tempestivamente ai nuovi dispositivi medici.
Per ulteriori chiarimenti sulla procedura speciale 510(k) della FDA, reach Freyr , un esperto in materia di regolamentazione. Rimanete informati. Rimanete conformi.