Panoramica dei servizi di conformità agli EU MDR
Il Regolamento UE sui dispositivi medici (MDR) è entrato in vigore il 26 maggio 2021, dopo una fase di transizione di 3 anni e un'ulteriore proroga di un anno a causa della pandemia COVID-19. I dispositivi che vengono lanciati sul mercato dell'UE devono ora essere conformi a queste normative e devono essere certificati CE secondo la EU MDR dagli organismi notificati accreditati secondo queste normative. I dispositivi che sono già stati certificati CE secondo la MDD dell'UE, tuttavia, hanno un periodo di tolleranza prima di dover soddisfare pienamente i requisiti della EU MDR . Durante questo periodo di tolleranza, i dispositivi certificati sia secondo la EU MDD che secondo la EU MDR co sul mercato con lo stesso status e senza essere soggetti a discriminazioni. Freyr offre servizi impareggiabili di conformità alla EU MDR per aiutare le aziende di dispositivi medici a soddisfare i requisiti EU MDR in modo tempestivo.
Calendario della transizione e nuove classificazioni dei dispositivi
Il Regolamento Europeo sui Dispositivi Medici (MDR) sarà pienamente efficace in tutti i Member States dell'EU e negli Stati dell'Associazione Europea di Libero Scambio (EFTA) a partire da maggio 2021 e fornisce ai produttori un periodo di transizione di 4 anni per la Certificazione EU MDR completa.
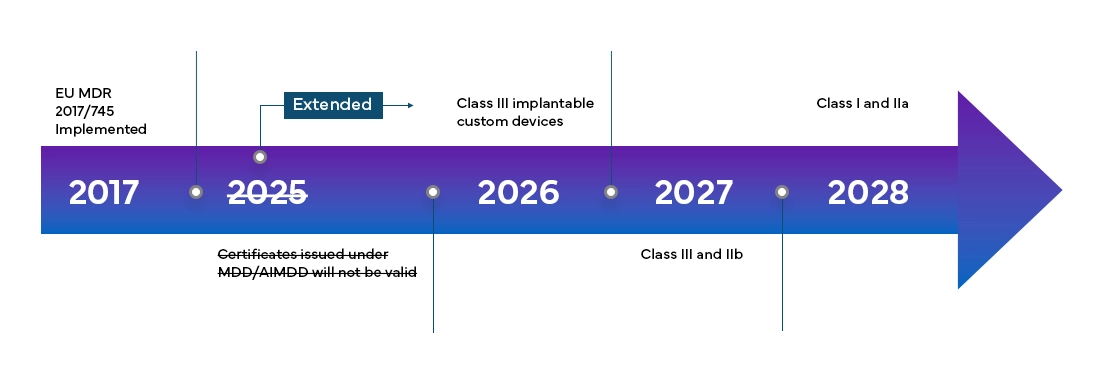
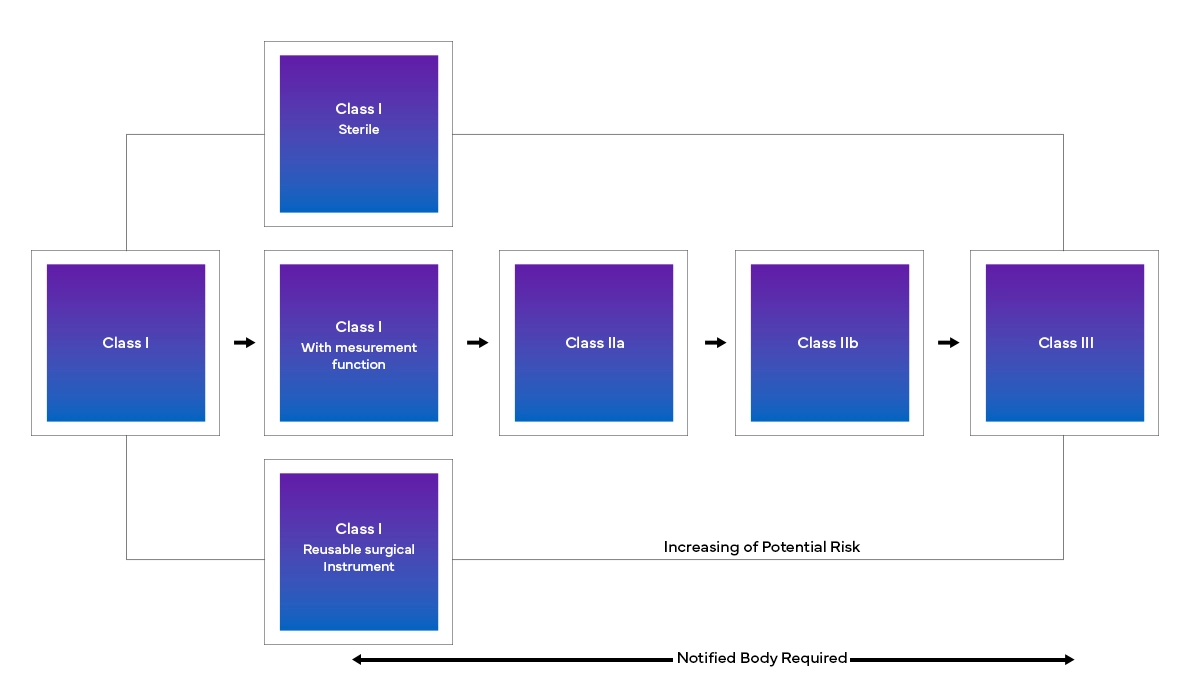
Il nuovo European Medical Device Regulation (MDR), come osservato, ha apportato modifiche anche al sistema di classificazione dei dispositivi esistenti, come ad esempio:
Dall'identificazione delle modifiche esatte da apportare alla loro implementazione in tempo reale, i produttori potrebbero dover affrontare una serie di sfide per conformarsi ai requisiti dell'EU MDR. Dalla decodifica della nuova struttura, alla classificazione accurata di un dispositivo, alla raccolta e presentazione di tutti i dati, sarà necessario un approccio normativo più dettagliato e trasversale per i produttori, per far fronte ai nuovi Regolamenti Europei sui Dispositivi Medici. Con una rigorosa analisi delle lacune, Freyr assiste i clienti con lo status quo e fornisce le azioni normative necessarie per la transizione e la conformità all'EU MDR.
Ottenete una consulenza esperta sulla vostra conformità all EU MDR
Servizi di conformità agli EU MDR
- Sviluppo di una chiara strategia di implementazione del regolamento sui dispositivi medici (MDR)
- Comprendere la nuova legislazione, condurre la Gap Analysis degli attuali sistemi di gestione della qualità (QMS) e dei processi in atto.
- Sviluppare un piano dettagliato con un approccio interfunzionale per determinare gli aspetti del sistema di qualità che dovranno essere modificati in conformità con il nuovo regolamento UE sui dispositivi medici.
- Formazione di più team per l'analisi dell'ambito del prodotto, la classificazione, la gestione del SGQ e così via, all'interno dell'organizzazione, con un unico punto di contatto in ciascun team.
- Allocazione e pianificazione delle risorse
- Considerare l'interazione del vostro SGQ con altre normative e utilizzare questa opportunità per snellire i processi, consentendo al contempo la flessibilità necessaria per incorporare cambiamenti futuri.
- Analizzare i dati dei test in atto e verificare eventuali requisiti aggiuntivi che MDR pone in essere
- Coordinamento delle aspettative e del piano di transizione con gli organismi notificati dell'UE
- Analisi delle lacune per i dispositivi medici esistenti dalla normativa UE MDD alla normativa EU MDR
- Supporto End-to-End per lo sviluppo del Clinical Evaluation Report (CER), inclusa la ricerca bibliografica secondo le linee guida del Regolamento Europeo sui Dispositivi Medici (EU MDR).
- Servizi End-to-End per i rapporti di sorveglianza post-commercializzazione (PMSR), il rapporto periodico di aggiornamento sulla sicurezza (PSUR) e la sintesi sulla sicurezza e la performance clinica (SSCP)
- Aumento delle risorse normative con opzioni di implementazione sia onshore che offshore
- Servizi del Rappresentante Autorizzato Europeo (EAR)
- Conformità MDR e assistenza alla presentazione agli organismi notificati
- Intelligence normativa che copre il processo di importazione di diversi mercati regolamentati
- Conformità del SGQ e simulazioni di audit
- Sistema e strumento di gestione documentale per le aziende MDR
- Classificazione e riclassificazione dei dispositivi in base al rischio
- Implementazione e consulenza UDI
- Servizi di sorveglianza post-market conformi al regolamento sui dispositivi medici dell'UE
- Gestione del rischio Consulenza ISO 14971
- Formazione interna e online
- Responsabile dei servizi e dell'assistenza in materia di conformità normativa
- Identificazione degli organismi notificati MDR

Per un supporto normativo end-to-end sull' EU MDR, contatta Freyr