Valutazione delle prestazioni dei dispositivi medici - Panoramica
Il mercato del Software as a Medical Device (SaMD) è in forte espansione a livello globale, grazie a fattori quali la crescente domanda di servizi sanitari a distanza e l'imminente innovazione nel settore della sanità digitale. In Europa, si prevede che il mercato SaMD crescerà del 27,1% circa nel periodo 2021-2027.
SaMD comprende un dispositivo medico diagnostico in vitro (IVD). È fondamentale sottolineare che i regolamenti dell'UE non utilizzano il termineSoftware as a Medical Device SaMD)". Utilizza invece il termine "Software per dispositivi medici", abbreviato in MDSW.
Nell'Unione Europea (UE), i SaMD sono disciplinati dal Regolamento sui dispositivi medici (MDR) 2017/745 e dal Regolamento sui diagnostici in vitro (IVDR) 2017/746. Questi regolamenti forniscono un quadro di riferimento per garantire la sicurezza e le prestazioni dei dispositivi medici, compresi i SaMD, nel mercato dell'UE.
Come si qualifica un software come dispositivo medico?
Per determinare se il vostro dispositivo è considerato SaMD dall'UE, dovete valutare se il software è destinato a essere utilizzato per uno (01) o più scopi medici senza essere parte di un dispositivo medico hardware. Se il software funziona da solo per soddisfare uno scopo medico, potrebbe essere qualificato come SaMD.
Tuttavia, se è destinato a pilotare un dispositivo medico hardware o è parte integrante di esso, non è considerato un software autonomo e quindi non è SaMD. Assicuratevi sempre che il vostro prodotto sia conforme alle più recenti normative e linee guida dell'UE per i dispositivi medici.
Come registrare il vostro SaMD nell'UE?
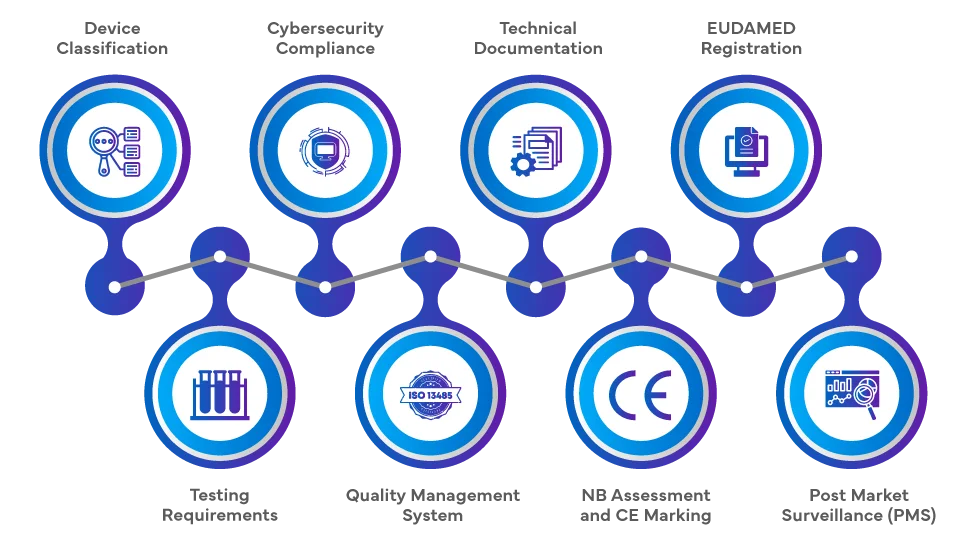
Il percorso normativo per l'introduzione di SaMD nel mercato dell'UE ai sensi del EU MDR 2017/745 e del EU IVDR 2017/746 comporta una serie completa di passaggi. I produttori devono classificare il prodotto software in base al suo scopo e livello di rischio, seguito da una valutazione di conformità, che può coinvolgere un Organismo Notificato (NB) per le classi a rischio più elevato. La documentazione tecnica, la valutazione clinica e l'aderenza ai Sistemi di Gestione della Qualità (QMS) sono fondamentali. Dopo una valutazione positiva, viene apposta la marcatura CE ed è necessaria la registrazione nella Banca Dati Europea dei Dispositivi Medici (EUDAMED). La continua Post-market Surveillance (PMS), la vigilanza e l'aderenza ai requisiti di cybersecurity completano il percorso normativo.
Domande frequenti (FAQ)
Un certificato EU MDR è un documento rilasciato da un NB designato a seguito di una valutazione di conformità ai sensi del Regolamento sui dispositivi medici dell'Unione EuropeaEU MDR 2017/745) o del Regolamento sui diagnostici in vitro (IVDR) 2017/746. Questo certificato verifica che un dispositivo medico soddisfi i requisiti stabiliti dal EU MDR, che comprende standard di sicurezza, prestazioni e qualità. Il certificato è necessario affinché i dispositivi medici/diagnostici in vitro possano essere legalmente commercializzati all'interno dell'UE.
Nell'UE, i dispositivi medici sono classificati in quattro (04) classi di rischio: Classe I, Classe IIa, Classe IIb e Classe III. La Classe I rappresenta i dispositivi con il rischio più basso, mentre la Classe III comprende i dispositivi con il rischio più elevato. La classificazione tiene conto di fattori quali l'invasività del dispositivo, la durata dell'uso, la parte del corpo interessata e i rischi potenziali associati alla progettazione tecnica e alla fabbricazione del dispositivo.
Allo stesso modo, le SaMD sono classificate in base al rischio che comportano per i pazienti e gli utenti, da basso rischio (Classe I) ad alto rischio (Classe III).
Il termineSoftware as a Medical Device SaMD)" si applica quando il software è progettato per uno (01) o più scopi medici, svolgendo queste funzioni in modo indipendente senza essere parte integrante di un dispositivo medico hardware. Il termine "Software in un dispositivo medico (SiMD)" viene invece utilizzato quando il software è integrato in un'apparecchiatura medica, noto come software incorporato o firmware.
È fondamentale sottolineare che i regolamenti dell'UE non utilizzano il termineSoftware as a Medical Device". Utilizza invece il termine "Software per dispositivi medici", abbreviato in MDSW.
"Software per dispositivi medici (MDSW)" si riferisce al software destinato a essere utilizzato, da solo o in combinazione, per uno scopo delineato nella definizione di "dispositivo medico" ai sensi delle EU MDR 2017/745 e UE IVDR 2017/746.
Il tempo necessario per ottenere l'approvazione di un SaMD nell'UE può variare significativamente in base a diversi fattori. Tra i fattori determinanti vi sono la classificazione del dispositivo, la complessità, il percorso di valutazione della conformità, il coinvolgimento di un ente normativo e l'efficienza del processo di presentazione della domanda.
Registrazione SaMD negli altri mercati (UE/Australia/Corea) per i dispositivi medici
- Strategia regolatoria EU MDR completa EU MDR per i SaMD.
- Supporto normativo e di market intelligence.
- Servizi di classificazione e registrazione dei prodotti per i SaMD.
- Supporto normativo per i documenti di sviluppo dei prodotti SaMD .
- Servizi di consulenza sugli studi di valutazione clinica SaMD .
- Gestione delle modifiche post-approvazione.
- Servizi EAR/UKRP/CH-Rep.

- Presentazioni di successo per varie classi di SaMD.
- Personale dedicato ed esperto per fornire supporto normativo.
- Presentazione puntuale dei prodotti.
- Accesso di affiliati locali per rispondere alle sfide dell Health Authority (HA) e ai requisiti specifici della lingua.
- Supporto all'interno del Paese o del Rappresentante Legale (LR), con un modello economicamente vantaggioso.
- Servizi di gestione delle risorse normative/aumento del personale.