Che cos'è l'etichettatura regolamentare?
Ultimo aggiornamento il: Agosto 2024
L'etichettatura regolatoria farmaceutica comporta la creazione, la revisione e la gestione di documenti critici che comunicano le informazioni essenziali sul prodotto agli stakeholder, garantendo la conformità agli standard regolatori globali. I componenti principali includono il Core Data SheetCDS) e il Company Core Data SheetCCDS), ricavati da fonti quali brochure sperimentali e dati post-marketing. Questo processo è fondamentale per trasmettere le informazioni sulla sicurezza e l'efficacia sulle etichette specifiche dei singoli Paesi e per allinearsi ai requisiti delle Health Authority (HA) .
Con un'attenzione particolare all'armonizzazione globale, l'etichettatura regolatoria farmaceutica risponde alle esigenze regolatorie in continua evoluzione, che comprendono le autorizzazioni per i nuovi prodotti, le autorizzazioni HA, le post-approvazioni e la gestione del ciclo di vita. La precisione e l'aderenza alle linee guida in evoluzione sono fondamentali per il successo dell'etichettatura regolatoria farmaceutica, in quanto influenzano l'autorizzazione al mercato, il profilo di sicurezza e la sostenibilità regolatoria complessiva di un prodotto.

Freyr, leader nei servizi di etichettatura normativa End-to-End per farmaci, vanta un team dedicato di oltre 180 esperti globali di etichettatura, eccellendo nella redazione di documenti cruciali come le Brochure per Sperimentatori (IB), i Fogli Dati Core di Sviluppo e le Informazioni Core di Sicurezza di Sviluppo. L'utilizzo dell'intelligenza artificiale migliora l'accuratezza e velocizza l'implementazione e la revisione dei fogli dati. Con un modello CCDS semplificato e processi basati sulla precisione, i servizi completi di Freyr soddisfano efficacemente le esigenze dinamiche dell'industria farmaceutica, fornendo un supporto impareggiabile per la conformità dell'etichettatura e il successo normativo.
Perché l'etichettatura regolamentare è importante nel settore farmaceutico?
- Garantire la sicurezza del paziente e la comunicazione delle informazioni: L'etichettatura regolamentare è fondamentale per la sicurezza del paziente. Le etichette forniscono informazioni essenziali sull'uso dei farmaci, sui dosaggi, sugli effetti collaterali e sulle controindicazioni. I pazienti, i medici prescrittori, gli operatori sanitari e gli assistenti si affidano a queste etichette per prendere decisioni informate. Un'etichettatura chiara e accurata riduce il rischio di errori farmacologici, eventi avversi e uso improprio.
Assicura che i pazienti ricevano il trattamento giusto e ne comprendano l'uso corretto. Inoltre, le autorità regolatorie richiedono che ogni prodotto farmaceutico in commercio abbia un'etichettatura che comunichi efficacemente le informazioni sul trattamento. - Conformità e mitigazione del rischio: La conformità alle normative sull'etichettatura non è solo una formalità; è un requisito legale. Organismi di regolamentazione come la US Food and Drug Administration (FDA), l'European Medicines Agency (EMA) e altri impongono un'etichettatura accurata e completa. La non conformità può comportare multe normative, danni alla reputazione del marchio e persino interruzioni temporanee della linea di produzione. Le aziende farmaceutiche devono dimostrare che i loro processi, metodi, test e attrezzature di etichettatura sono in grado di produrre costantemente prodotti sicuri ed efficaci. Un'etichettatura correttamente validata mitiga i rischi e garantisce l'adesione alle buone pratiche di fabbricazione (GMP).
- Accesso al mercato e armonizzazione globale: Etichette ben strutturate facilitano l'accesso al mercato globale. Un'etichettatura coerente tra le varie regioni snellisce i processi, riduce le ridondanze e si allinea agli standard armonizzati. Con l'adozione da parte delle autorità di regolamentazione internazionali dei requisiti di convalida delle GMP, tra cui la serializzazione, le catene di fornitura farmaceutiche si trovano ad affrontare una crescente complessità. Le aziende che danno priorità alla conformità dell'etichettatura creano fiducia, migliorano l'accettazione del mercato e si posizionano per il successo in un panorama competitivo.
Qual è il processo di approvazione dell'etichettatura?
Il processo di approvazione dell'etichettatura nell'industria farmaceutica prevede diverse fasi per garantire che tutte le informazioni relative ai farmaci siano accurate, conformi e chiare sia per gli operatori sanitari che per i pazienti. Si inizia con la stesura del contenuto dell'etichetta, che include dettagli su dosaggio, somministrazione, sicurezza e avvertenze. I team medici e regolatori rivedono poi internamente questa bozza per assicurarsi che sia in linea con gli standard regolatori locali e internazionali. Una volta finalizzata, l'etichetta viene presentata alle autorità sanitarie per l'approvazione, dove viene sottoposta a un rigoroso esame per verificarne la conformità ai requisiti di sicurezza ed efficacia. Solo dopo aver ricevuto l'approvazione ufficiale, l'etichetta può essere utilizzata per la commercializzazione del farmaco.
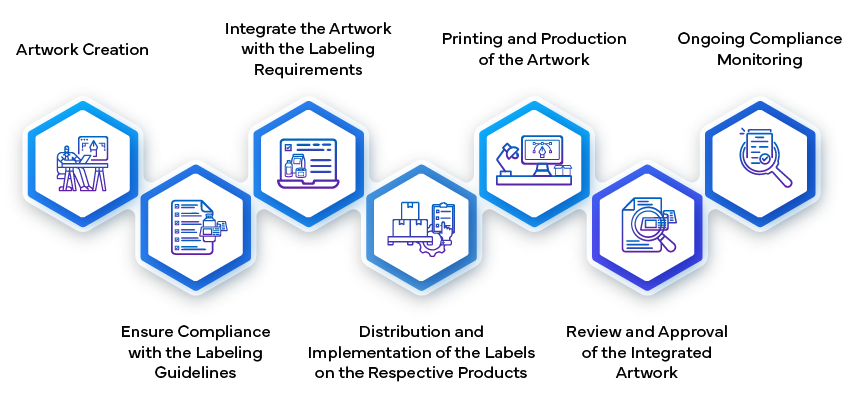
- Creazione e invio dell'etichetta:
- Raccolta iniziale dei dati: Le aziende farmaceutiche raccolgono tutti i dati rilevanti per l'etichetta del farmaco. Questi includono informazioni su efficacia, sicurezza, dosaggi, indicazioni, controindicazioni e rischi potenziali. L'etichetta è uno strumento di comunicazione fondamentale per gli operatori sanitari e i pazienti.
- Presentazione alle Autorità Regolatorie: L'azienda presenta i dati dell'etichetta agli organismi di regolamentazione, come la US Food and Drug Administration (FDA) o l'European Medicines Agency (EMA). Queste agenzie valutano i dati per garantire che il farmaco offra benefici che superano i suoi rischi noti e potenziali per la popolazione target.
- Analisi contestuale: I revisori analizzano la condizione o la malattia a cui il farmaco è destinato. Considerano il panorama terapeutico esistente, soppesando i rischi del farmaco rispetto ai suoi benefici. Per esempio, un farmaco che tratta una malattia pericolosa per la vita e che non ha una terapia alternativa può essere approvato anche se i rischi sarebbero inaccettabili per una patologia non pericolosa per la vita.
- Valutazione dei dati clinici: I revisori dell FDA valutano le informazioni sui benefici e sui rischi clinici presentate dal produttore del farmaco. Tengono conto delle incertezze derivanti da dati imperfetti o incompleti. In genere, l'agenzia si aspetta i risultati di studi clinici ben progettati per convalidare l'efficacia e la sicurezza del farmaco.
- Stabilire l'Artwork:
- Una volta approvato il contenuto dell'etichetta, il passo successivo è la creazione della grafica dell'etichetta. Ciò implica la progettazione degli elementi visivi, del layout, dei caratteri e della grafica. La grafica deve essere conforme alle linee guida normative e rappresentare accuratamente le informazioni sull'etichetta.
- artwork dell'etichetta artwork revisioni interne all'azienda farmaceutica per garantirne la coerenza e la conformità. Include dettagli quali istruzioni di dosaggio, avvertenze, condizioni di conservazione e informazioni di contatto.
- artwork definitivo artwork sottoposto all'approvazione delle autorità regolatorie. Questa fase garantisce che la presentazione visiva dell'etichetta sia conforme agli standard di qualità e comunichi in modo efficace le informazioni fondamentali agli utenti.
- Produzione e implementazione:
- Una volta approvata, la grafica dell'etichetta diventa parte integrante del confezionamento del farmaco. I produttori si assicurano che le etichette siano correttamente applicate a ogni unità di prodotto (ad esempio, flaconi, blister, fiale).
- Le procedure di controllo della qualità verificano che le etichette siano conformi alle specifiche, compresi i contenuti accurati, la leggibilità e l'aderenza alle linee guida del design.
- L'etichetta funge da ponte tra l'azienda farmaceutica, le agenzie regolatorie, gli operatori sanitari e i pazienti. Svolge un ruolo fondamentale nel garantire un uso sicuro ed efficace dei farmaci durante l'intero ciclo di vita del prodotto.
Quali sono le sfide più comuni nell'etichettatura normativa?
Le sfide più comuni nell'etichettatura regolatoria includono la necessità di tenere il passo con l'evoluzione dei requisiti regolatori, la gestione di un'etichettatura multilingue e la garanzia di coerenza tra diversi portafogli di prodotti. La natura dinamica degli standard normativi per l'etichettatura farmaceutica richiede una continua vigilanza per rimanere aggiornati sui requisiti più recenti. Adattarsi ai cambiamenti normativi e implementare tempestivamente gli aggiornamenti necessari al contenuto e al formato delle etichette è essenziale per mantenere la conformità e garantire la sicurezza dei pazienti.
| Sfida | Descrizione |
|---|---|
| Requisiti normativi in continua evoluzione | Affrontare la continua evoluzione delle normative e delle linee guida, che richiede un costante aggiornamento della documentazione di etichettatura. |
| Armonizzazione globale | Garantire la coerenza delle informazioni sui prodotti in diverse regioni, allineandosi ai vari requisiti delle diverse autorità sanitarie. |
| Integrazione dei dati post-marketing | Gestire l'incorporazione dei dati di sicurezza ed efficacia post-marketing nell'etichettatura, mantenendone l'accuratezza e la pertinenza. |
| Conformità agli standard di etichettatura locali | Aderire agli standard di etichettatura specifici dei singoli Paesi, tenendo conto delle variazioni linguistiche, culturali e di formattazione. |
| Gestione efficiente delle modifiche all'etichettatura | Semplificare i processi per tracciare, implementare e documentare le modifiche all'etichettatura in modo tempestivo e accurato. |
L'etichettatura multilingue rappresenta una sfida significativa per le aziende farmaceutiche che operano nei mercati globali. Tradurre accuratamente i contenuti dell'etichettatura in più lingue, rispettando le sfumature linguistiche e normative regionali, richiede attenzione ai dettagli e solidi processi di gestione delle traduzioni. Garantire coerenza e chiarezza tra le diverse versioni linguistiche è fondamentale per comunicare efficacemente informazioni vitali a popolazioni di pazienti diverse.
Mantenere la coerenza tra i portafogli di prodotti rappresenta un'altra sfida comune nell'etichettatura regolatoria. Le aziende farmaceutiche spesso gestiscono diversi prodotti con requisiti di etichettatura, formulazioni e indicazioni differenti. Per ottenere coerenza e conformità tra le diverse linee di prodotti, soddisfacendo al contempo i requisiti normativi specifici per ciascun prodotto, è necessario disporre di processi e sistemi efficienti che garantiscano l'uniformità dei contenuti, del formato e della messaggistica dell'etichettatura.
Quali sono le principali normative che regolano l'etichettatura dei prodotti farmaceutici?
L'etichettatura dei farmaci è regolata da una serie complessa di norme volte a garantire la sicurezza, l'efficacia e l'uso accurato dei farmaci. Di seguito ne elenchiamo alcune:
FDA US FDA Food and Drug Administration degli Stati Uniti)
FDA US FDA l'etichettatura dei prodotti farmaceutici attraverso una serie di norme rigorose delineate nel Codice dei regolamenti federali (CFR) Titolo 21. Tali norme richiedono che le etichette forniscano informazioni complete, comprese le indicazioni terapeutiche, le istruzioni per l'uso, le controindicazioni e i potenziali effetti collaterali. La FDA l'importanza di un linguaggio chiaro, preciso e inequivocabile per garantire la sicurezza dei pazienti e un processo decisionale informato da parte degli operatori sanitari. Inoltre, i requisiti di etichettatura FDA si estendono a vari aspetti quali imballaggi, foglietti illustrativi ed etichettatura elettronica, garantendo che tutte le informazioni siano accessibili e standardizzate in diversi formati. La conformità a queste norme è obbligatoria per l'approvazione dei farmaci e la loro permanenza sul mercato negli Stati Uniti.
EMA (Agenzia europea per i medicinali)
L'EMA supervisiona l'etichettatura dei farmaci nell'Unione Europea attraverso direttive e linee guida volte ad armonizzare l'etichettatura tra gli member states. La Direttiva 2001/83/CE European Commission è il fulcro di questi sforzi e specifica i requisiti per il Riassunto delle Caratteristiche del ProdottoSmPC), i foglietti illustrativi per i pazienti e le etichette delle confezioni. L'EMA garantisce che l'etichettatura contenga informazioni essenziali sia per gli operatori sanitari che per i pazienti, promuovendo un uso sicuro ed efficace dei farmaci in tutta l'EU (European Union). Inoltre, l'etichettatura deve essere disponibile nelle lingue ufficiali degli member states in cui il farmaco è commercializzato, a testimonianza dell'impegno dell'EMA per l'accessibilità e la centralità del paziente.
TGA (Therapeutic Goods Administration)
In Australia, il TGA è responsabile della regolamentazione dell'etichettatura dei farmaci ai sensi del Therapeutic Goods Act 1989. Le linee guida del TGA prevedono che le etichette dei farmaci forniscano informazioni chiare, accurate e complete sul prodotto, compresi gli ingredienti, le indicazioni, il dosaggio e i potenziali rischi. I requisiti di etichettatura sono concepiti per proteggere la salute pubblica, garantendo che i consumatori e gli operatori sanitari dispongano delle informazioni necessarie per utilizzare i farmaci in modo sicuro ed efficace. Il TGA pone inoltre un'enfasi significativa sulla leggibilità delle etichette, richiedendo che siano scritte in un inglese semplice e che le informazioni critiche siano visualizzate in modo evidente per prevenire l'uso improprio e gli errori di medicazione.
Health Canada
Health Canada regolamenta l'etichettatura dei farmaci attraverso un quadro che dà priorità alla sicurezza e al benessere dei pazienti e degli operatori sanitari. Il Food and Drugs Act e i relativi regolamenti definiscono i requisiti per le etichette dei farmaci, che devono includere informazioni dettagliate sulla composizione del prodotto, le indicazioni, le controindicazioni e i potenziali effetti collaterali. Health Canada richiede anche che le etichette siano bilingue, cioè presentate sia in inglese che in francese, per tenere conto della diversità linguistica del Paese. Inoltre, Health Canada aggiorna regolarmente i requisiti di etichettatura per riflettere le nuove evidenze scientifiche e l'evoluzione delle esigenze di salute pubblica, assicurando che l'etichettatura rimanga pertinente ed efficace nel promuovere un uso sicuro dei farmaci.
PMDA (Agenzia per i prodotti farmaceutici e i dispositivi medici)
La PMDA, l'autorità regolatoria giapponese, vigila sull'etichettatura dei farmaci in conformità alla legge sugli affari farmaceutici e alle relative linee guida. La PMDA richiede che le etichette dei farmaci forniscano informazioni complete, comprese le indicazioni, le istruzioni per il dosaggio e i potenziali effetti avversi, in un formato facilmente comprensibile sia per gli operatori sanitari che per i pazienti. Il PMDA richiede inoltre che le etichette includano avvertenze e precauzioni specifiche per la popolazione giapponese, tenendo conto di fattori quali le differenze genetiche e le pratiche culturali. Questo approccio garantisce che i farmaci siano utilizzati in modo sicuro ed efficace in Giappone, con un'etichettatura adattata alle esigenze specifiche del mercato locale.
NMPA (Amministrazione nazionale dei prodotti medici)
In Cina, l'NMPA disciplina l'etichettatura dei farmaci attraverso un quadro normativo che enfatizza l'accuratezza, la chiarezza e la sicurezza. La legge sull'amministrazione dei farmaci della Repubblica Popolare Cinese definisce i requisiti per le etichette dei farmaci, che devono includere informazioni su indicazioni, dosaggio, controindicazioni e potenziali effetti collaterali. L'NMPA richiede anche che le etichette siano presentate in cinese semplificato per garantire l'accessibilità alla popolazione locale. Inoltre, l'NMPA prevede che le etichette siano sottoposte a una revisione rigorosa durante il processo di approvazione dei farmaci, per garantire la conformità agli standard nazionali e proteggere la salute pubblica prevenendo gli errori e l'uso improprio dei farmaci.
In che modo un partner normativo può aiutare a raggiungere la conformità ai requisiti di etichettatura?
Un partner normativo è fondamentale per raggiungere la conformità ai requisiti di etichettatura, offrendo competenze specialistiche e supporto completo. Guida le aziende attraverso il complesso panorama normativo, garantendo che i materiali di etichettatura — inclusi imballaggi, inserti ed etichette elettroniche — aderiscano ai requisiti specifici delle diverse autorità sanitarie come US FDA, EMA, TGA, Health Canada, PMDA e NMPA. Ciò implica la comprensione e l'applicazione delle ultime normative, che possono variare significativamente tra le regioni, per garantire che tutte le informazioni sul prodotto siano accurate, complete e conformi.
Inoltre, un partner normativo aiuta a snellire il processo di etichettatura fornendo servizi fondamentali come la creazione, la revisione e la convalida dei contenuti. Essi assistono nella stesura e nella revisione dei contenuti dell'etichettatura per allinearsi agli standard normativi e garantire che siano incluse tutte le informazioni necessarie, dagli elenchi degli ingredienti alle istruzioni d'uso, dalle avvertenze di sicurezza alle condizioni di conservazione. In questo modo si riduce il rischio di errori e omissioni che potrebbero causare ritardi normativi o ritiri dal mercato, accelerando il time-to-market dei nuovi prodotti.

Inoltre, un partner normativo supporta le aziende nel mantenimento della conformità, monitorando gli aggiornamenti normativi e implementando le modifiche necessarie. Offrono consulenza strategica per adattare le etichette alle nuove linee guida o ai requisiti dei mercati emergenti, aiutando le aziende a evitare problemi di non conformità e garantendo che i loro prodotti rimangano in linea con le normative vigenti. Sfruttando la propria esperienza e rimanendo al passo con i cambiamenti normativi, un partner normativo aiuta le aziende a navigare nel dinamico panorama dell'etichettatura in modo efficiente ed efficace.
Come possono le aziende iniziare con i Regulatory Labeling Services?
Per iniziare a usufruire dei Regulatory labeling services, le aziende devono innanzitutto valutare le loro specifiche esigenze di etichettatura in base ai mercati di destinazione e ai requisiti normativi. Quindi, devono collaborare con un fornitore di servizi normativi affidabile, esperto in standard di etichettatura globali. Questo fornitore può assistere nella stesura, nella revisione e nell'aggiornamento delle etichette per garantire la conformità. Inoltre, l'implementazione di un sistema centralizzato di gestione delle etichette aiuta a snellire il processo, garantendo la coerenza di tutte le etichette dei prodotti. Le revisioni e gli aggiornamenti periodici sono essenziali per mantenere le etichette allineate alle normative in evoluzione.
I Regulatory Labeling Services possono aiutare nel monitoraggio post-vendita?
Sì, i Regulatory labeling services possono effettivamente contribuire al monitoraggio post-vendita dei prodotti farmaceutici. Questi servizi svolgono un ruolo cruciale nel supportare la sorveglianza post-vendita, facilitando la gestione degli aggiornamenti dell'etichettatura, gestendo le modifiche all'etichettatura relative alla sicurezza e garantendo la conformità ai requisiti normativi post-approvazione. Mantenendo informazioni accurate e aggiornate sull'etichettatura, i Regulatory labeling services aiutano le aziende farmaceutiche a rispondere ai problemi di sicurezza e a implementare tempestivamente le modifiche necessarie per sostenere la conformità regolatoria e la sicurezza dei pazienti.
Inoltre, i Regulatory labeling services possono contribuire alla diffusione efficiente di informazioni aggiornate sulla sicurezza agli operatori sanitari e ai pazienti. In caso di nuove scoperte sulla sicurezza o di cambiamenti nei profili di rischio dei prodotti farmaceutici, gli esperti di etichettatura regolatoria possono aiutare ad aggiornare rapidamente il contenuto dell'etichetta per riflettere i più recenti dati sulla sicurezza e i requisiti regolatori. Questo approccio proattivo al monitoraggio post-market e agli aggiornamenti delle etichette favorisce la comunicazione tempestiva di importanti informazioni sulla sicurezza agli operatori sanitari e ai pazienti, contribuendo a migliorare la farmacovigilanza e la cura dei pazienti.
In generale, i Regulatory labeling services forniscono un valido supporto al monitoraggio post-market, garantendo che i prodotti farmaceutici mantengano un'etichettatura accurata e conforme per tutto il loro ciclo di vita. Sfruttando l'esperienza di professionisti del settore regolatorio e processi efficienti di gestione dell'etichettatura, le aziende possono affrontare efficacemente le considerazioni sulla sicurezza post-market e gli obblighi regolatori, contribuendo così alla sicurezza e all'efficacia dei loro prodotti sul mercato.
Perché scegliere Freyr?
![us di noi]()
Un decennio di eccellenza nell'etichettatura regolatoria
![us di noi]()
Oltre 180 esperti globali in servizi di etichettatura
![us di noi]()
Specializzato nella creazione e nella gestione di documenti essenziali
![us di noi]()
Esperienza in brochure sperimentali, schede tecniche di base e schede tecniche di base aziendali.
![us di noi]()
Impegno per la conformità e la precisione a livello globale
![us di noi]()
Utilizza l'AI (Artificial Intelligence) per una navigazione efficiente della Normativa






Fatti rapidi








Domande frequenti
Le schede tecniche di baseCDS) forniscono un riepilogo consolidato delle informazioni critiche sui farmaci, tra cui indicazioni, dosaggi e profili di sicurezza. Assicurano una comunicazione coerente dei dettagli essenziali in tutti i mercati globali, facilitando la conformità alle normative e un processo decisionale informato. CDS servono anche come riferimento per la creazione delle etichette dei prodotti locali.
Le Investigational Brochures (IB) descrivono in dettaglio i dati degli studi clinici e le informazioni sullo sviluppo dei farmaci per uso sperimentale, mentre le Company Core Data Sheets (CCDS) riassumono i dati chiave di sicurezza ed efficacia per scopi normativi globali, guidando il contenuto e gli aggiornamenti delle etichette. Le CCDS sono utilizzate per creare etichette specifiche per prodotto per l'approvazione all'immissione in commercio.
L'intelligenza artificiale migliora l'etichettatura normativa automatizzando l'analisi dei dati, migliorando l'accuratezza nella creazione dei contenuti e accelerando la revisione dei documenti. Gli strumenti di intelligenza artificiale semplificano i processi di etichettatura e garantiscono la coerenza tra i diversi requisiti normativi. Inoltre, aiutano a prevedere e ad affrontare potenziali problemi di conformità.
L'etichettatura multilingue garantisce che i prodotti farmaceutici siano accessibili a popolazioni di pazienti diverse, soddisfacendo i requisiti normativi regionali e migliorando la sicurezza grazie a istruzioni e avvertenze chiare e comprensibili in più lingue. Ciò riduce il rischio di errori di comunicazione e di somministrazione dei farmaci.
Un sistema centralizzato di gestione delle etichette coordina la creazione, la revisione e l'aggiornamento dei documenti di etichettatura, garantendo coerenza e conformità nei mercati globali. Semplifica i processi e mantiene informazioni accurate e aggiornate sui prodotti. Questo sistema supporta anche una gestione efficiente delle modifiche all'etichettatura e degli aggiornamenti normativi.
Structured Product Labeling (SPL) è un formato basato su XML utilizzato per l'etichettatura dei farmaci che standardizza e organizza le informazioni sul prodotto. Garantisce coerenza e facilita lo scambio di dati tra le agenzie di regolamentazione e i produttori. SPL supporta la gestione efficiente delle informazioni di Labelling durante l'intero ciclo di vita di un prodotto.
Il Global Location Number (GLN) è un identificatore unico utilizzato per identificare le sedi e le entità all'interno della catena di fornitura. Aiuta a tracciare e gestire con precisione i prodotti farmaceutici nei mercati globali. I GLN garantiscono una distribuzione precisa ed efficiente dei prodotti e una gestione delle scorte.
Il National Drug Code (NDC) è un identificativo unico per i farmaci, assegnato dalla FDA. Aiuta a identificare con precisione i prodotti farmaceutici e facilita la gestione e la tracciabilità dell'inventario. L'NDC è fondamentale per un'accurata dispensazione dei farmaci e per la stesura dei rapporti normativi.
Il dossier per lo sperimentatoreIB) fornisce informazioni dettagliate sui dati clinici e preclinici di un farmaco in sperimentazione. Viene utilizzato per informare gli sperimentatori sulla sicurezza, l'efficacia e il dosaggio del farmaco ai fini dello studio. Il IB supporta inoltre un processo decisionale etico e informato nella ricerca clinica.




