
4 minuti di lettura
Con l'attuazione da parte US and Drug Administration (USFDA) della normativa definitiva relativa al sistema di gestione della qualità (QMSR) nel 2024, produttori di dispositivi medici produttori adeguarsi alle modifiche per commercializzare e distribuire i propri dispositivi sul mercato statunitense.
Questa norma aggiorna le Quality System Regulations (QSR) USFDAallineandosi alla ISO 13485:2016, che è lo standard internazionale per i sistemi di gestione della qualità dei dispositivi medici. produttori di dispositivi medici produttori un periodo di transizione di due anni per conformarsi, il che rende necessario per le organizzazioni adattarsi ai nuovi requisiti per evitare la non conformità al momento dell'ispezione.
Che cos'è il QMSR?
FDA è un approccio semplificato ai requisiti QMS, che costituisce un aggiornamento della precedente struttura QSR. Questo allineamento è fondamentale perché semplifica la conformità globale per i produttori quelli che operano a livello mondiale. Questa armonizzazione consentirà alle aziende di soddisfare i requisiti normativi sia negli US in altri mercati in modo molto più uniforme.
Il QMSR impone miglioramenti nella gestione del rischio, nella progettazione dei dispositivi e nella sorveglianza post-commercializzazione. Questo ruolo può aggiungere maggiore complessità, ma offre anche ai produttori la possibilità di standardizzare le loro procedure di qualità, il che a sua volta aumenta la sicurezza del dispositivo e migliora la documentazione, elementi vitali durante le ispezioni USFDA.
Cambiamenti chiave nel QMSR
- Armonizzazione con ISO 13485: Questo è il passo più critico che consente ai produttori di dispositivi medici di adottare standard accettati a livello internazionale. La USFDA ha riconosciuto che molte aziende di dispositivi medici sono già conformi a ISO 13485, il che riduce gli sforzi duplicati.
- La gestione del rischio enfatizza la gestione del rischio per l'intero ciclo di vita del dispositivo medico. I produttori di dispositivi medici devono dimostrare una gestione e un controllo del rischio efficaci.
- Progettazione e controlli del dispositivo: Nell'ambito del QMSR, i controlli di progettazione sono stati estesi per garantire che i produttori di dispositivi medici tengano pienamente conto delle esigenze dell'utente, della sicurezza del dispositivo e dei criteri di prestazione, che è un'area di interesse durante l'ispezione USFDA.
- Sorveglianza Post-Commercializzazione: Le aziende devono migliorare il sistema di monitoraggio post-commercializzazione. Ciò richiederà ai produttori di raccogliere informazioni sulla sicurezza e l'efficacia del dispositivo, il che aiuta a individuare e identificare rapidamente i problemi.
- Documentazione e tenuta dei registri: Questa è la norma finale che pone l'accento sulla documentazione. Un'accurata e corretta registrazione è fondamentale durante l'ispezione.
Passi per prepararsi all'ispezione della US Food and Drug Administration:
Con l'ispezione che durerà fino al 2026, le aziende hanno due anni di tempo per i loro sistemi di qualità, in linea con il QMSR. Tuttavia, aspettare l'ultimo minuto può essere rischioso.
Procedura per USFDA nell'ambito del quadro QMSR per i settori in cui l'attuale QMS non si basa sul QSR:
- Condurre un'analisi delle lacune: Questa è la prima fase in cui il sistema di qualità attuale diverge dai nuovi requisiti del QMSR. Un'analisi approfondita delle lacune aiuterà a identificare le aree che necessitano di aggiornamenti, come la gestione del rischio, la sorveglianza post-vendita e i controlli di progettazione.
- Aggiornare la procedura di gestione del rischio: Assicurarsi che le attività di gestione del rischio siano abbinate all'intero ciclo di vita del prodotto, dalla progettazione alla sorveglianza post-vendita.
- Rivedere il controllo della progettazione: I produttori dovrebbero assicurarsi che il processo di progettazione sia solido e ben documentato. Convalidare che il processo di progettazione sia solido, ben documentato e pienamente integrato nel vostro sistema di gestione della qualità.
- Migliorare la sorveglianza post-commercializzazione: Implementare sistemi di monitoraggio delle prestazioni dei dispositivi dopo la loro immissione sul mercato. Ciò potrebbe comportare la creazione di meccanismi di feedback da parte dei clienti, la raccolta di dati clinici e il loro monitoraggio.
- Formazione e documentazione: formazione del personale sui nuovi requisiti, in particolare di coloro che sono coinvolti nella gestione della qualità e nella conformità normativa. Ciò garantisce che tutti i processi di documentazione siano in linea con le aspettative del QMSR.
- Certificazione di terze parti: se la vostra azienda non è ISO 13485 , questo potrebbe essere il momento giusto per prenderla in considerazione. Ottenere ISO 13485 può darvi un vantaggio nel soddisfare i requisiti della USFDA aumentare la credibilità nei mercati globali.
Come superare il periodo di transizione di due anni
Per sfruttare al meglio il periodo di transizione e garantire la conformità al Regolamento sul Sistema di Gestione della Qualità (QMSR) USFDA, produttori adottare un approccio proattivo. Ecco una roadmap suggerita:
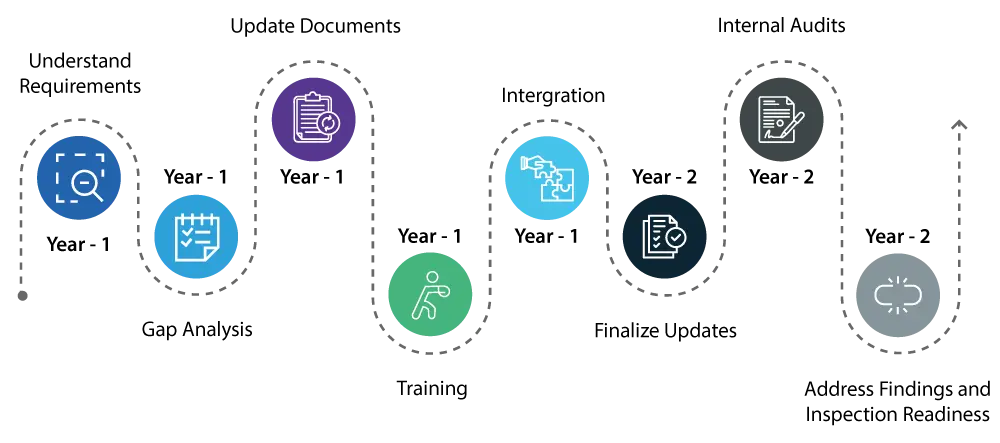
Anno 1:
- Comprendere i requisiti: Iniziare a comprendere a fondo i cambiamenti introdotti dal QMSR. Ciò implica un esame dettagliato dei nuovi requisiti e delle loro differenze rispetto all'attuale regolamento sul Sistema Qualità (SQ).
- Conoscere le lacune: condurre un'analisi completa delle lacune per identificare le aree del vostro attuale sistema di qualità che richiedono aggiornamenti per soddisfare i nuovi standard QMSR.
- Aggiornamento e correzione dei documenti: avviare gli aggiornamenti necessari al sistema di qualità, concentrandosi su aree quali la gestione del rischio e i controlli di progettazione, che sono componenti critici del QMSR.
- Formazione: iniziate a formare il vostro personale sulle nuove norme per garantire che tutti i soggetti coinvolti siano consapevoli dei cambiamenti e comprendano il proprio ruolo nel mantenimento della conformità.
- Integrazione: iniziare a integrare i nuovi requisiti QMSR nelle operazioni quotidiane per rendere più agevole la transizione.
Anno 2:
- Proseguire con l'implementazione delle modifiche al sistema di qualità, assicurandosi che tutti gli aggiornamenti siano pienamente integrati e operativi.
- Condurre audit interni approfonditi per verificare che gli aggiornamenti siano efficaci e che il sistema di qualità sia pienamente allineato ai requisiti del QMSR.
- Affrontare tempestivamente qualsiasi risultato degli audit interni per garantire la conformità di tutti gli aspetti del sistema di qualità.
- Entro la fine del secondo anno, il vostro sistema di qualità dovrebbe essere pienamente conforme al QMSR e dovreste essere pronti per USFDA con la certezza che non ci saranno problemi rilevanti.
Seguendo questa roadmap, i produttori possono non solo soddisfare i requisiti della USFDA, ma anche stabilire un sistema di qualità robusto che sia efficiente, standardizzato e riconosciuto a livello globale. Questo approccio proattivo contribuirà a garantire una transizione agevole alle nuove normative e a mantenere i più alti standard di qualità e sicurezza per i dispositivi medici.
Conclusione: Prendere misure proattive
Prepararsi alle ispezioni USFDA secondo la nuova norma QMSR non significa solo evitare sanzioni, ma migliorare la sicurezza e l'efficacia dei dispositivi. Armonizzandosi con la ISO 13485, la USFDA sta definendo aspettative più elevate, ma anche fornendo un percorso verso una conformità globale più snella. I produttori che iniziano ad adattarsi precocemente dovrebbero concentrarsi su aree chiave come la gestione del rischio e la sorveglianza post-commercializzazione e assicurarsi che i loro sistemi di qualità siano aggiornati e robusti, che non solo soddisferanno le aspettative normative, ma miglioreranno anche il loro vantaggio competitivo sul mercato.
