
5 minuti di lettura
Il Medical Device Single Audit Program MDSAP) consente a un organismo di audit riconosciuto (AO) di condurre un unico audit del Quality Management System (QMS) di un produttore di dispositivi medici. Fornisce i requisiti normativi pertinenti per cinque paesi, ovvero Brasile (ANVISA), Stati Uniti (FDA), Giappone (PMDA), Canada (Health Canada) e Australia (TGA). Oltre alle autorità di regolamentazione partecipanti, diversi altri partner internazionali (gli osservatori ufficiali e i membri affiliati) sono coinvolti nel MDSAP.
La certificazione MDSAP è obbligatoria per Health Canada per i dispositivi di Classe II, III e IV, ma è volontaria per gli altri quattro paesi. Ha promosso la trasparenza e l'allineamento normativo tra le autorità partecipanti e ha minimizzato la necessità di audit multipli, risparmiando così tempo e risorse dei produttori di dispositivi medici. Per darti una migliore prospettiva sul programma MDSAP, qui abbiamo cercato di rispondere alle quindici domande più frequenti.
- Perché è stato sviluppato il programma MDSAP quando esiste una certificazione ISO 13485 accettata a livello globale?
L'MDSAP è stato sviluppato per ridurre l'onere degli audit normativi per i produttori di dispositivi medici e per promuovere un maggiore allineamento degli approcci normativi e dei requisiti tecnici basati su standard internazionali e migliori pratiche. Si concentra sul portare coerenza, prevedibilità e trasparenza ai programmi normativi standardizzando le procedure e le pratiche dei regolatori e delle organizzazioni di audit di terze parti.
L'audit si basa sui requisiti del sistema di gestione della qualità (QMS) previsti dalla norma ISO 13485 sui requisiti normativi del Paese partecipante in cui saranno commercializzati i dispositivi medici.
- Quali sono i criteri di idoneità per sottoporsi a un MDSAP ?
Qualsiasi fabbricante di dispositivi medici che intenda commercializzare il proprio dispositivo nei paesi partecipanti può sottoporsi a un audit MDSAP. Tuttavia, ogni Autorità Regolatoria può stabilire criteri di esclusione per determinate condizioni, se necessario.
Ad esempio, in Giappone, le eccezioni per l'ammissibilità sono:
- Un sito di produzione registrato (RMS) che produce dispositivi medici a base di tessuti umani/animali.
- Un RMS che produce IVD radioattivi, e
- L'istituzione di un titolare dell'autorizzazione all'immissione in commercio (MAH)
- MDSAP include i prodotti combinati?
I dispositivi medici che includono farmaci (sostanze medicinali) o prodotti biologici (ad esempio, materiali di origine animale resi non vitali, tessuti, cellule o sostanze di origine microbica o ricombinante, sangue umano o estratti di sangue umano o emoderivati, ecc.) sono considerati prodotti combinati e possono essere inclusi nell'ambito di un MDSAP .
Tuttavia, a causa delle differenze nella regolamentazione di questi prodotti nelle giurisdizioni delle autorità di regolamentazione partecipanti, i rapporti MDSAP e i documenti di certificazione potrebbero non essere considerati un'alternativa ai requisiti di ispezione e valutazione in alcune giurisdizioni.
Australia- I prodotti combinati sono soggetti a un esame a distanza della TGA nell'ambito della Valutazione di Conformità australiana. Ma un audit MDSAP efficace può ridurre le ispezioni per questi dispositivi.
Brasile, Giappone- I prodotti combinati considerati dispositivi medici sono inclusi nel MDSAP, poiché non ci sono requisiti specifici riguardo al Sistema di Gestione della Qualità.
Canada- Il modello MDSAP copre i requisiti QMS per i prodotti combinati considerati dispositivi medici.
US- Gli audit MDSAP non sono considerati alternative alle ispezioni FDA per i prodotti combinati.
- Posso selezionare il Paese oggetto dell MDSAP ?
Sì, l'audit viene eseguito in base all'ambito dichiarato nella domanda di servizi di certificazione. produttori di dispositivi medici produttori tenuti a rispettare le normative solo nelle giurisdizioni in cui i loro prodotti saranno commercializzati.
- Sono un produttore di dispositivi medici dagli US, intenzionato a commercializzare il mio dispositivo solo in Giappone. Sto per sottopormi a un audit MDSAP. Devo conformarmi anche ai requisiti di altri paesi?
No, i produttori di dispositivi medici sono tenuti a conformarsi ai requisiti e alle normative ISO 13485 solo nelle giurisdizioni in cui i loro prodotti devono essere commercializzati.
- La mia Organizzazione di Audit (AO) e l'Organismo Notificato Europeo sono gli stessi. Posso essere sottoposto a audit per entrambi contemporaneamente?
Se l'AO e l'organismo notificato europeo sono gli stessi, la valutazione di conformità può essere eseguita dopo aver condotto MDSAP , non contemporaneamente. Gli organismi notificati europei sono osservatori per MDSAP e la valutazione di conformità viene condotta secondo EU MDR . Per MDSAP, la valutazione viene eseguita secondo i requisiti della norma ISO 13485 i requisiti normativi dei paesi partecipanti nell'ambito di applicazione.
- Qual è la differenza tra le valutazioni di fase I e II?
Il processo di audit MDSAP prevede due fasi. L'audit iniziale, chiamato anche audit di certificazione iniziale, consiste in audit di Fase I e Fase II.
L'audit di Fase I comprende l'esame della documentazione e la valutazione della disponibilità del fabbricante di dispositivi medici a sottoporsi a un audit di Fase II.
L'audit di fase II viene eseguito per verificare se tutti i requisiti applicabili della norma ISO 13485 gli altri requisiti normativi dell'autorità di regolamentazione nell'ambito di applicazione sono stati implementati.
- Quanti revisori posso aspettarmi per un MDSAP ?
Determinazione del tempo di audit specifica come determinare la durata dell'audit in loco in giorni/uomo. L'AO decide quanti auditor comporranno il team di audit. Ad esempio, un audit di (06) giorni/uomo può essere completato in tre (03) giorni da un team di due (02) auditor.
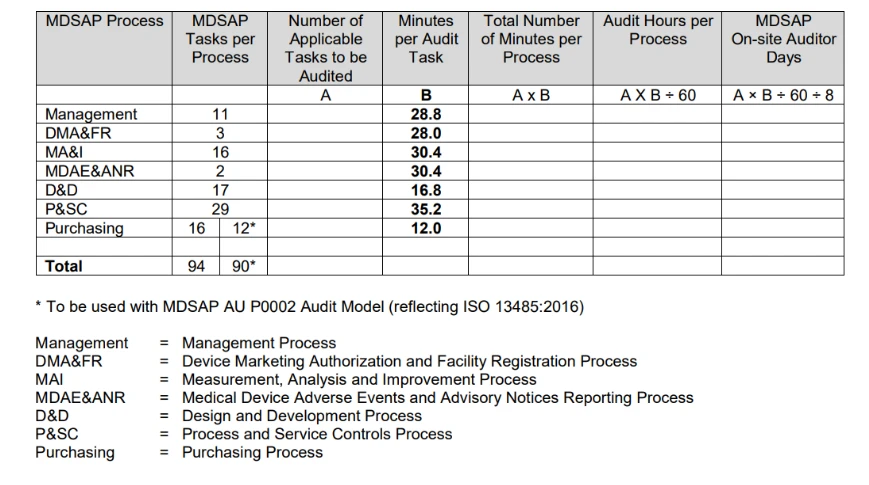
- Come viene programmato MDSAP ?
La Procedura di Determinazione del Tempo di Audit, emessa dalla FDA, riassume il processo per determinare la durata del calcolo dell'audit nella seguente tabella.
Il calcolo della durata dell'audit si basa principalmente sul numero di compiti di audit applicabili associati al tipo di audit da condurre e alle attività specifiche dell'organizzazione da verificare.
Per informazioni dettagliate al riguardo, è possibile fare riferimento a MDSAP .
- Esiste una guida o una checklist a cui posso accedere per garantire la conformità con un MDSAP ?
Sì, è possibile accedere al documento MDSAP Approach. Si tratta di una guida ben organizzata pubblicata dalla USFDA rimanda a sezioni specifiche della ISO 13485:2016 e alle normative pertinenti emanate dalla TGA australiana, ANVISA brasiliana, Health Canada canadese,PMDA giapponese e dallaFDA US .
- Qual è il ruolo di un osservatore in un MDSAP ?
Un osservatore MDSAP è un'Autorità Regolatoria a cui è consentito partecipare a riunioni, valutazioni e altre attività, ma che non utilizza i risultati MDSAP. Gli osservatori sono rappresentati nel Consiglio delle Autorità Regolatorie MDSAP (RAC) da un dirigente di alto livello.
- Quali sono i passi successivi da compiere se ho ricevuto un voto pari o superiore a 4?
Il sistema di classificazione viene assegnato alle non conformità osservate durante l'audit da AO. Un punteggio di 4 o 5 indica un rischio elevato di intervento. Per ogni non conformità registrata è necessario fornire un piano di ripristino entro 15 giorni di calendario dalla data di emissione del rapporto di non conformità. Il piano di rimedio deve includere i risultati dell'indagine sulla non conformità, le sue cause e le azioni correttive pianificate per evitare che si ripeta. L'evidenza dell'attuazione del piano/azione di rimedio deve essere fornita entro trenta (30) giorni di calendario dalla data di conclusione dell'audit.
- Esiste una differenza nel processo di approccio all'audit da parte di un revisore interno rispetto a un AO?
MDSAP un approccio basato sui processi. L'AO tenderà a esaminare i collegamenti e i fili conduttori, mentre un auditor interno potrebbe concentrarsi maggiormente su un aspetto funzionale alla volta. Pertanto, l'AO potrebbe individuare una non conformità in un'area funzionale e cercare risposte in un'area funzionale diversa. Tuttavia, seguire l'approccio basato sui processi potrebbe risultare dirompente durante un audit interno.
- Posso appellarmi all'AO se posso dimostrare che una non conformità registrata non è valida?
L'AO prevede un processo di appello o di contestazione, che può essere utilizzato se si riesce a dimostrare che una non conformità registrata non è valida. Tuttavia, i gradi assegnati alle non conformità non possono essere modificati a causa di azioni correttive. Possono essere modificati solo sulla base di prove che dimostrino che non erano validi.
- Quanto dura la validità del MDSAP ?
I produttori di dispositivi medici certificati nell'ambito del programma MDSAP saranno sottoposti a audit annuali, secondo un ciclo di certificazione triennale. L'audit iniziale è un audit completo del QMS del produttore di dispositivi medici. È seguito da audit di sorveglianza condotti annualmente per due anni consecutivi. Il ciclo ricomincia con un audit di ricertificazione nel terzo anno.
Per saperne di più sui nostri MDSAP , contatta Freyr oggi stesso per fissare una chiamata con i nostri esperti.









