
2 minuti di lettura
Dopo la scoperta iniziale di impurità di nitrosammine nei farmaci e negli ingredienti farmaceutici attivi (APIs) da parte della USFDA a metà del 2018, anche gli organismi di regolamentazione dell'UE si sono uniti a molti altri paesi nel tentativo di prevenire i rischi connessi. Hanno richiamato diversi medicinali che presentano rischi per la salute a causa della sostanza. Nota per le sue proprietà cancerogene, la nitrosammina può rivelarsi dannosa se ingerita al di sopra dei livelli accettati dagli esseri umani. La sostanza, N-nitrosodimetilammina (NDMA), ha un limite accettabile di 96 ng/giorno e qualsiasi quantità superiore a questo è considerata inaccettabile nei farmaci e negli APIs.
Le nitrosammine si formano quando ammine secondarie, terziarie o quaternarie reagiscono con un agente nitrosante. Tutti i farmaci che contengono principi attivi sintetizzati chimicamente vengono controllati per le impurità. Il Comitato per i Medicinali per Uso Umano (CHMP) ha esaminato e redatto un rapporto di valutazione. Ha chiesto ai Titolari di Autorizzazione all'Immissione in Commercio (AIC) di seguire le ultime linee guida per la revisione di tutti i medicinali chimici e biologici attualmente disponibili sul mercato per il consumo umano.
I MAH sono responsabili di assicurarsi che il processo di produzione di tutti i prodotti biologici e chimici sia revisionato periodicamente per identificare eventuali contaminazioni. Una volta identificate, devono essere intraprese le misure pertinenti per mitigare i rischi che esse comportano. Sebbene le probabilità di tali contaminazioni siano minori durante la produzione di farmaci chimici e biologici, l'Agenzia Europea per i Medicinali (EMA) non vuole correre rischi. Le aziende farmaceutiche devono assicurarsi che i protocolli di produzione pertinenti siano in atto secondo le più recenti linee guida dell'EMA. Sono anche responsabili di controllare i livelli di nitrosammine nei farmaci disponibili sul mercato e di mantenerli entro il limite accettabile.
Dopo aver finalizzato una revisione ai sensi dell'articolo 5, paragrafo 3, del Regolamento (CE) n. 726/2004 (richiesta di revisione) lo scorso anno, l'EMA ha emesso nuove linee guida per evitare la presenza di contaminazione da nitrosammine nei medicinali per uso umano. Il processo è lo stesso per i prodotti autorizzati a livello nazionale e per quelli autorizzati a livello centrale. L'EMA, insieme alla Direzione Europea per la Qualità dei Medicinali e dell'Assistenza Sanitaria (EDQM), implementerà l'articolo 5, paragrafo 3, del CHMP. Ecco il processo che dovrebbe essere seguito per rimanere conformi alle modifiche.
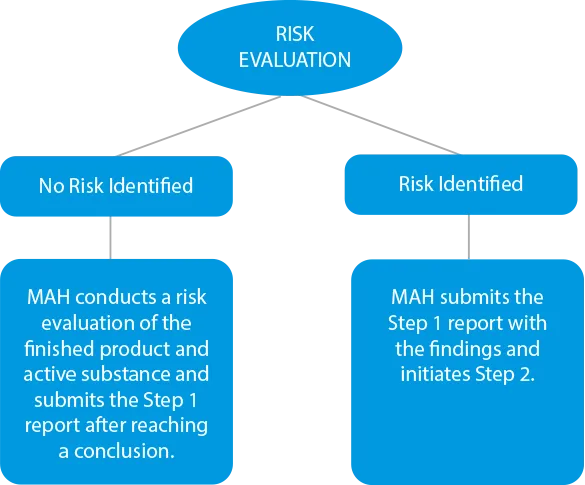
La guida in tre fasi per i MAH
- Valutazione del rischio I produttori devono condurre un processo di valutazione del rischio per identificare le sostanze attive e il prodotto finito al fine di verificare i livelli di nitrosammine. In caso di contaminazione incrociata, questa deve essere inclusa anche nel rapporto finale. Le scadenze per le presentazioni in questa fase sono state fissate al 31 marzo 2021 per i medicinali chimici e al 1° luglio 2021 per i farmaci biologici.
![]()
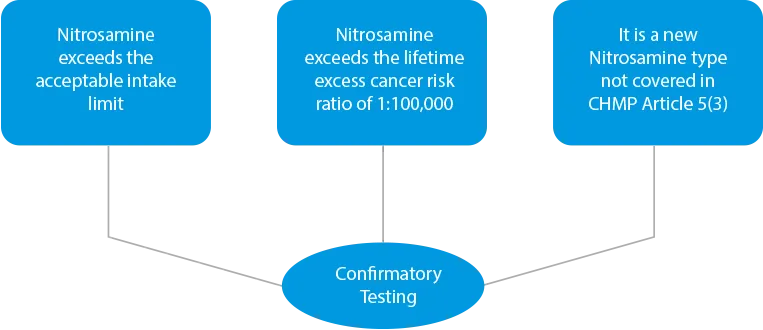
- Test di conferma - In caso di riscontro di contaminazione incrociata o se i prodotti vengono identificati come rischiosi a causa della presenza di livelli più elevati di nitrosammina, è necessario eseguire un test di conferma. I test di conferma sono obbligatori in tre (03) casi.
![]()
- Modifiche all'autorizzazione all'immissione in commercio – Quando viene rilevata la presenza di nitrosamina, devono essere eseguiti due test di conferma per comunicare le letture corrette all'EMA. Sulla base di questi, i MAH devono richiedere la modifica del processo di fabbricazione. Ciò viene fatto utilizzando le procedure normative standard con l'aiuto di una variazione al Modello di Autorizzazione all'Immissione in Commercio. Le tempistiche per questi sono il 26 settembre 2022 per i prodotti chimici e il 1° luglio 2023 per i medicinali biologici.
La strada ideale per i MAH per affrontare la nuova conformità dei livelli di nitrosammina
Poiché questo è l'ultimo sviluppo che l'EMA ha proposto per mitigare i rischi legati ai livelli di nitrosamina nei farmaci e a una situazione di contaminazione incrociata, l'intero processo può essere piuttosto impegnativo per i MAHs e i produttori di APIs. Sia che si tratti di presentare il Modello di Risposta quando viene rilevata una contaminazione al Passaggio 1 o di condurre test conseguenti, ogni fase deve essere conforme alle nuove regole. I MAHs devono collaborare con esperti normativi che siano aggiornati su tutti gli ultimi cambiamenti e garantire la conformità con la nuova guida. Scegli il partner giusto come Freyr per evitare ritardi ed errori.









