3 minuti di lettura
Il regolamento dell'Unione europea sui dispositivi medici (EU MDR) 2017/745 definisce l'ambito di applicazione dei dispositivi medici sterili come dispositivi che devono essere privi di microrganismi (batteri, virus, ecc.). Anche i dispositivi di classe I con funzione sterile possono ottenere un'autocertificazione, ma i requisiti sono diversi rispetto ad altre sottocategorie di dispositivi di classe I.
I requisiti relativi ai dispositivi di Classe I variano notevolmente dalla produzione all'immissione sul mercato. Lo stesso vale per i dispositivi medici di Classe I in condizioni sterili. Ad esempio, se si tratta di un dispositivo di Classe I con funzione sterile, i requisiti per la produzione e l'istituzione di un Quality Management System (QMS) molto diversi rispetto ai dispositivi di Classe I con funzioni di misurazione.
Questo articolo ha lo scopo di fornire una checklist concisa da tenere in considerazione quando si immette un dispositivo di Classe I in condizioni sterili nell'Unione Europea. La checklist aiuterà i produttori prendere decisioni informate su diversi aspetti.
Dos!
Conoscere l'ambito di applicazione prima della produzione
Secondo l'EU MDR, i dispositivi medici con funzioni sterili devono essere sviluppati e fabbricati con procedure appropriate per garantirne la sterilità fino alla loro immissione sul mercato. I dispositivi possono essere riutilizzati solo dopo appropriate procedure di pulizia, disinfezione e sterilizzazione.
Scelta delle corrette pratiche di sterilizzazione
Esistono diversi metodi di sterilizzazione tra cui un produttore può scegliere per sterilizzare il dispositivo. Il metodo con ossido di etilene (EtO) è ampiamente utilizzato per sterilizzare i dispositivi medici. La scelta del metodo di sterilizzazione deve essere adeguatamente giustificata. Alcune norme che produttori seguire per stabilire, garantire e mantenere la sterilizzazione sono EN ISO 14937, ISO TS 21387, ISO TS 13004, ISO 11137 e altre.
Inoltre, si noti che la documentazione del metodo di sterilizzazione dettagliato, del metodo in-process e della convalida dei dati deve essere eseguita con precisione. Le informazioni sulla sterilizzazione, come la temperatura di riscaldamento, le condizioni ambientali, le convalide del processo, ecc., devono essere fornite in conformità ai requisiti dell'EU MDR.
Conoscere i requisiti e le linee guida da seguire
Uno dei passaggi fondamentali consiste nel documentare i requisiti tecnici e conoscere gli standard accettati ai sensi EU MDR. Ad esempio, la norma EN 556 è lo standard europeo per i dispositivi sterilizzati. La norma EN 556 specifica i requisiti per designare un dispositivo medico come sterile. Inoltre, la norma ISO TS 19930 specifica le strategie per garantire la sterilità dei dispositivi. Tuttavia, non è ancora stata adottata come standard UE.
Conoscere il percorso
Per ottenere il marchio CE, è necessario informarsi sul coinvolgimento degli organismi notificati e sul percorso di valutazione della conformità da seguire. Inoltre, i dispositivi medici in condizioni di sterilità sono tenuti a seguire gli Allegati IX e XI.
Il coinvolgimento di un organismo notificato è necessario ma limitato. Nel caso dei dispositivi sterili, il coinvolgimento degli organismi notificati riguarderà il mantenimento e la garanzia delle condizioni di sterilità. Ad esempio, le valutazioni di conformità comporteranno valutazioni microbiologiche, metodi asettici, ecc.
Non farlo!
Non perdete i requisiti per l'imballaggio e l'etichettatura

Esistono ulteriori requisiti per la conformità dell'imballaggio e dell'etichettatura dei dispositivi sterili. Nel rispettare i requisiti di imballaggio ed etichettatura, non bisogna dimenticare le informazioni da apporre. Il primo e principale passo è l'apposizione del simbolo di sterilità (menzionato di seguito) sull'etichetta del dispositivo.
Devono essere incluse informazioni quali la dichiarazione di sterilità, il metodo di sterilizzazione, le istruzioni da seguire nel caso in cui la confezione sterile sia danneggiata o aperta involontariamente, ecc. Inoltre, è necessario includere la durata di conservazione del prodotto sterile e il processo di risterilizzazione (se applicabile). Sul dispositivo deve essere apposto anche il numero di identificazione di un organismo notificato.
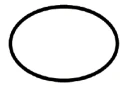
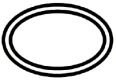
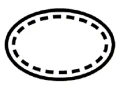
Alcuni dei simboli che devono essere aggiunti (a seconda dell'applicabilità) nell'etichettatura dei dispositivi medici sterili sono:
Simbolo | Indicazione |
| Dispositivi medici sterili con metodo di sterilizzazione utilizzato come tecnica asettica |
| Dispositivi medici sterili con metodo di sterilizzazione utilizzato come ossido di etilene |
| Dispositivi medici sterili con metodo di sterilizzazione utilizzato come calore secco o vapore |
| Dispositivi medici sterili con metodo di sterilizzazione utilizzato come perossido di idrogeno vaporizzato |
| Dispositivi medici sterili con metodo di sterilizzazione utilizzato come radiazione |
| Il dispositivo non può essere sterilizzato nuovamente |
| Il dispositivo non è sterilizzato |
| Non utilizzarlo se la confezione è danneggiata |
| Sistema di barriera sterile singolo |
| Sistema a doppia barriera sterile |
| Sistema di barriera sterile singolo con confezione protettiva interna |
| Sistema di barriera sterile singolo con imballaggio protettivo esterno |


Non dimenticate i requisiti per il collocamento successivo alla commercializzazione
I dispositivi sterili sono tenuti a mantenere una registrazione degli eventi avversi in caso di eventi avversi. In questo caso, gli eventi registrati, come i documenti, devono essere esibiti dalle autorità competenti su richiesta.
L'EU MDR ha requisiti specifici per quanto riguarda l'autocertificazione. Si tratta di una procedura complessa, il fattore principale essendo le diverse classi all'interno del dispositivo di Classe I e i loro diversi aspetti dei requisiti per ottenere un marchio CE. I produttori con dispositivi di Classe I in condizioni sterili dovrebbero essere consapevoli della designazione dell'ambito e dei requisiti specifici.
Desiderate comprendere nel dettaglio i vari aspetti dei dispositivi sterili di classe I? Vorreste discuterne con un esperto comprovato? us.









