
2 minuti di lettura
FDA US FDA pubblicato un documento guida che aiuterà il settore e il personale dell'Agenzia sanitaria (HA) a determinare quando una modifica software a un dispositivo medico richiede al produttore di presentare e ottenere FDA per una nuova notifica pre-commercializzazione (510(k)). Questa guida intende migliorare la prevedibilità, la coerenza e la trasparenza del processo decisionale relativo alla "tempistica di presentazione", fornendo un approccio meno oneroso e descrivendo il quadro normativo, le politiche e le pratiche alla base di tale decisione, in particolare per quanto riguarda le modifiche al software. Scopriamo nel dettaglio la FDA .
Principi FDA e diagramma di flusso FDA
Con l'intento di assistere i produttori di dispositivi medici produttori dei principi fondamentali, il documento fornisce un diagramma di flusso, ulteriori chiarimenti ed esempi necessari per prendere decisioni in merito a una nuova notifica pre-commercializzazione 510(k) per una modifica software apportata a un dispositivo già approvato negli US. Inoltre, è necessario seguire diversi principi guida durante l'utilizzo di questa guida per determinare se presentare una nuova 510(k) per modificare un dispositivo esistente. Alcuni di essi sono ampiamente noti e derivano dall'attuale politica FDA (k), mentre altri sono necessari per utilizzare lo schema logico menzionato nella presente guida. Secondo la guida, lo schema fornito non può coprire tutte le possibili complessità relative a tali modifiche e il modo in cui queste influenzano la decisione. Pertanto, per determinare la necessità di una nuova notifica pre-commercializzazione 510(k), produttori di dispositivi medici produttori considerare i principi generali e il diagramma di flusso riassunti di seguito.
- Modifiche che influiscono sulla sicurezza o sull'efficacia di un dispositivo
- Valutazione iniziale basata sul rischio
- Conseguenze indesiderate dei cambiamenti
- Utilizzo della gestione del rischio
- Ruolo dei test (attività di verifica e convalida) nel valutare se una modifica possa influire significativamente sulla sicurezza e sull'efficacia.
- Valutare le modifiche simultanee per determinare se è necessaria la presentazione di un nuovo 510(k).
- Dispositivo comparativo appropriato ed effetto cumulativo dei cambiamenti
- Requisiti del documento (21 CFR Parte 820)
- Presentazione di 510(k) per dispositivi modificati
- Determinazioni di equivalenza sostanziale
- È probabile che la presentazione di un nuovo 510(k) sia necessaria se un produttore modifica il proprio dispositivo per influenzarne la sicurezza o l'efficacia. Tuttavia, le modifiche che non sono destinate a influire sulla sicurezza o sull'efficacia del dispositivo devono essere valutate.
- Per determinare se una modifica o un cambiamento potrebbe influire significativamente sulla sicurezza o sull'efficacia, il fabbricante deve innanzitutto condurre una valutazione basata sul rischio per sapere se la modifica potrebbe influire positivamente o negativamente sulla sicurezza o sull'efficacia del dispositivo. Questa valutazione basata sul rischio deve identificare e analizzare tutti i nuovi rischi e le variazioni dei rischi esistenti derivanti dalla modifica del dispositivo e portare a una decisione iniziale sulla necessità di presentare una nuova domanda 510(k).
- A volte ci sono ulteriori conseguenze non intenzionali o non pianificate che possono essere innescate durante la presentazione del software. Il diagramma di flusso deve valutare queste conseguenze per determinare se è necessaria la presentazione di una nuova 510(k).
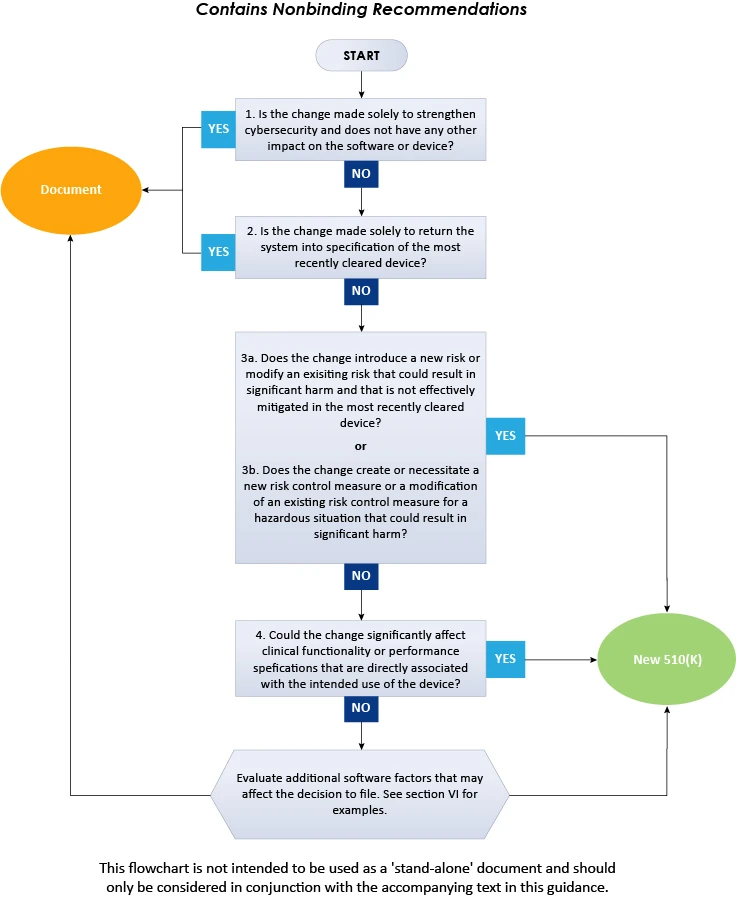
Il diagramma di flusso sopra riportato illustra una procedura dettagliata da seguire per decidere in merito alla presentazione della domanda 510(k) per le modifiche software nei dispositivi esistenti. In conclusione, le attuali FDA descrivono in dettaglio l'approccio che i produttori di dispositivi medici devono seguire produttori decidere se le modifiche software apportate a un dispositivo medico esistente richiedono la presentazione di una nuova domanda 510(k). Per ulteriori approfondimenti sulle FDA , consultare Freyr, un esperto comprovato in materia di regolamentazione. Rimanete informati. Rimanete conformi.









