
3 minuti di lettura
La gestione del rischio è un'attività critica in tutte le fasi del ciclo di vita dei dispositivi medici, poiché influisce direttamente sulla sicurezza e sul benessere dei pazienti. I rischi sono inevitabili, ma possono essere ridotti se le aziende sono consapevoli dei pericoli incombenti e seguono procedure efficaci di gestione del rischio.
L'analisi dei modi di guasto e degli effetti (FMEA) è uno strumento di revisione per identificare possibili guasti in un progetto, in un processo di produzione o di assemblaggio, o in un prodotto o servizio di un dispositivo. Le "modalità di guasto" si riferiscono ai modi in cui un dispositivo potrebbe guastarsi, con potenziali conseguenze per i pazienti. "L'analisi degli effetti si riferisce all'esame delle conseguenze di tali guasti. Si tratta di un approccio graduale per garantire l'affidabilità e la qualità di un dispositivo.
Esistono due tipi di FMEA: FMEA di Progettazione (DFMEA) e FMEA di Processo (PFMEA). Nel contesto dei dispositivi medici, i produttori di dispositivi utilizzano la DFMEA per valutare i guasti relativi alla progettazione e alle specifiche del dispositivo, mentre la PFMEA è utilizzata per migliorare il processo di produzione.
Sebbene la FMEA riguardi l'aspetto del rischio, non è un sistema di gestione del rischio. I requisiti della gestione del rischio sono definiti dalla ISO 14971:2019, che serve come quadro di riferimento per i produttori di dispositivi medici per prevedere la probabilità dei rischi e le loro conseguenze durante l'intero ciclo di vita del prodotto. La metodologia FMEA di valutazione del rischio non è allineata con la ISO 14971:2019. La FMEA ha un proprio standard accettato a livello internazionale, la IEC 60812:2018, che spiega come l'analisi dei modi e degli effetti dei guasti viene pianificata, eseguita, documentata e mantenuta. La FMEA e la ISO 14971 differiscono l'una dall'altra in alcuni aspetti, che sono i seguenti:
Uso normale e condizioni di guasto
Secondo la ISO 14971, la gestione del rischio include sia l'uso normale che quello scorretto del dispositivo, mentre l'FMEA include i rischi associati solo al guasto del dispositivo. Un semplice esempio di ciò sarebbero i rischi associati alla linea endovenosa (IV). La ISO 14971 considera il rischio potenziale di infezione nonostante la corretta somministrazione di un IV. Ciò può essere dovuto a varie ragioni, come la bassa immunità del paziente e le infezioni presenti nell'ambiente ospedaliero/clinico. Questi rischi non sono presi in considerazione in una valutazione FMEA. Sebbene i produttori di dispositivi medici non possano evitare completamente questi rischi, possono rendere gli utenti consapevoli dei rischi residui associati all'uso del dispositivo.
Valutazione della gravità
La ISO 14971 considera la gravità del rischio in base al danno alla vita delle persone, mentre l'FMEA la considera in base alle anomalie nelle prestazioni del sistema. La gravità del rischio può essere considerata bassa nell'FMEA se si verifica una perdita di funzione minore, anche se può portare alla perdita di vite umane. La gravità sarà considerata alta se il dispositivo si rompe.
Ad esempio, FDA un filo guida (Classe I) destinato ad essere inserito all'interno di un catetere percutaneo per guidare il catetere attraverso un vaso sanguigno. Il filo guida in questione presenta il rischio che il rivestimento si sfaldi. La FMEA ha potenzialmente classificato questo rischio come di bassa gravità dopo la valutazione, ma può avere gravi implicazioni per la salute del paziente.
Procedura di valutazione del rischio/modalità di guasto
La FMEA e la ISO 14971 differiscono nel modo in cui viene valutato il rischio. Nell'FMEA, il rischio viene valutato identificando le modalità e gli effetti di guasto potenziali, quindi classificando la gravità dei guasti. Ciascuna delle cause potenziali viene identificata e ne viene determinata la probabilità di accadimento. Il rischio viene valutato in base al numero di priorità del rischio (RPN).
Mappatura FMEA
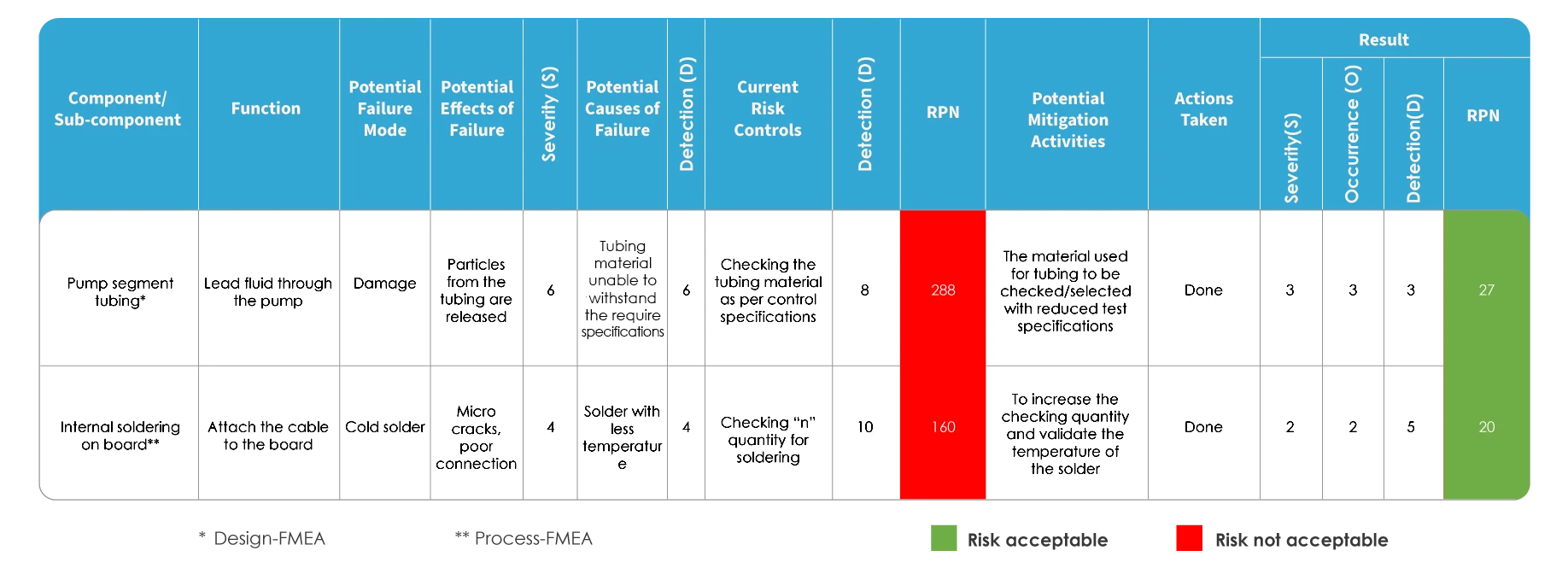
Nel caso della mappatura della gestione del rischio secondo la norma ISO 14971, viene utilizzato uno strumento di tracciabilità noto come Hazard Traceability Matrix (HTM). Essa comprende l'analisi, la valutazione, il controllo e la valutazione del rischio residuo.
Matrice di tracciabilità dei pericoli
| Analisi del rischio | Il rischio Eval. | Controllo del rischio | ||||||||||||
| ID | Pericolo | Sequenza o combinazione di eventi ragionevolmente prevedibile | Pericoloso Situazione | Harm | Occasione | Gravità | Accettabile? | Opzioni di controllo del rischio e motivazioni | Controllo del rischio Misure | Il rischio Controllo Verifica | Stato | Potenziale di rischio | Gravità del rischio | Rischio residuo |
| 1 | Tensione di linea | L'utente utilizza il dispositivo | L'utente/paziente può essere esposto alla tensione di rete mentre è a contatto con il dispositivo. | Morte dell'utente/paziente | 5 | 5 | N | La sicurezza può essere garantita da una modifica del progetto e dalle misure di protezione che possono essere posizionate in loco. | Progettazione conforme alla norma IEC 61010. | I test elettrici devono essere eseguiti in conformità alla norma IEC 61010. | Fatto | 2 | 2 | Y |
![]()
Avendo discusso le differenze tra i due, si può concludere che ISO 14971 segue un approccio completo alla gestione del rischio, mentre FMEA è più uno strumento di affidabilità. Tuttavia, i produttori di dispositivi medici dovrebbero conformarsi a ISO 14971 per soddisfare le aspettative delle autorità normative sugli standard di gestione del rischio.
Per saperne di più sulla conformità alla norma ISO 14971:2016 e sui servizi di consulenza in materia di gestione dei rischi, contatta Freyr oggi stesso!









