3 minuti di lettura
L'etichettatura è parte integrante della commercializzazione dei dispositivi medici. L'etichetta è un'informazione apposta sul dispositivo e/o sulla confezione in un formato leggibile dall'uomo. Lo scopo principale dell'etichettatura è fornire informazioni di sicurezza agli utenti, che possono essere operatori sanitari, consumatori o qualsiasi altra persona interessata.
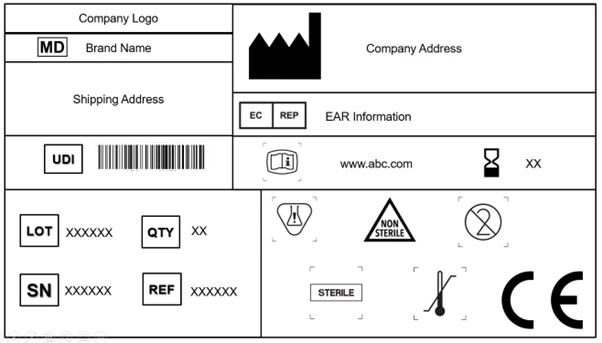
Tutte le autorità di regolamentazione globali hanno determinati requisiti di etichettatura. Allo stesso modo, l'UE ha specificato i requisiti di etichettatura nel Capitolo III dell'Allegato I del Regolamento UE sui dispositivi medici (EU MDR) 2017/745. La cosa più importante da notare è includere tutti i simboli che coprono le informazioni richieste nell'etichettatura del dispositivo e nei documenti (opuscoli, manuali, IFU, ecc.) che lo accompagnano.
Alcune delle considerazioni critiche relative all'etichettatura da tenere presenti ai fini della conformità al EU MDR sono:
1. Simbologia di etichettatura dei dispositivi medici
Ogni produttore è tenuto a incorporare il simbolo del dispositivo medico, che indica che il prodotto fornito al mercato dell'UE è un dispositivo medico. È obbligatorio apporre questo simbolo sul dispositivo e su tutti i livelli di imballaggio. Inoltre, l'etichetta deve riportare il nome commerciale e il nome originale del dispositivo.
2. Dispositivi speciali
Se il prodotto è un dispositivo speciale o personalizzato, il suo stato deve essere indicato sull'etichetta. Ad esempio, se il prodotto è destinato esclusivamente a indagini cliniche, l'etichetta deve indicarlo esplicitamente.
Per i dispositivi con materiali assorbenti o che possono disperdersi localmente nel corpo umano, l'etichettatura deve indicare la composizione del materiale e i dettagli quantitativi sui costituenti principali.
Anche l'etichettatura esplicita è richiesta nel caso di dispositivi monouso e sterili. Per i dispositivi ricondizionati, l'etichettatura deve indicare il numero di volte in cui possono essere ricondizionati, il numero di volte in cui sono stati ricondizionati finora e il metodo di sterilizzazione utilizzato.
3. Presenza di sostanze tossiche
La dichiarazione della presenza di sostanze CMR (cancerogene, mutagene, tossiche per la riproduzione) e di sostanze che alterano il sistema endocrino è obbligatoria sulle etichette se la concentrazione è superiore allo 0,1% in peso/peso. L'elenco di tali sostanze deve essere apposto sul dispositivo e/o sull'imballaggio.
Inoltre, sui dispositivi deve essere apposta un'etichetta sulla presenza di derivati del sangue e dei tessuti (anche se contenuti nella sostanza medicinale del dispositivo combinato).
4. Standard armonizzati
Il regolamento EU MDR riconosce e accetta la norma ISO 15223-1: 2021. Il documento stabilisce i simboli da utilizzare nell'etichettatura dei dispositivi medici e dei loro imballaggi. Il capitolo 3 (23.1,h) dell'allegato I EU MDR che è possibile utilizzare simboli riconosciuti a livello internazionale e, nel caso di regioni in cui tali simboli non sono riconosciuti, è necessario fornire la descrizione degli stessi in un documento allegato al dispositivo.
5. UDI
Gli articoli 27, 28, 29 e l'Allegato VI (A, B, C) definiscono nel dettaglio le norme e i regolamenti per l'UDI. L'etichetta deve ora contenere un vettore UDI [rappresentazione dell'identificazione automatica per la cattura dei dati (AIDC) e dell'interpretazione a lettura umana (HRI) dell'UDI] sul dispositivo e sui livelli di imballaggio superiori. L'imballaggio superiore del dispositivo (escluse le confezioni di spedizione) avrà un proprio supporto UDI.
6. Informazioni elettroniche per l'uso (eIFU)
L'indirizzo web (URL) sotto forma di eIFU può anche essere inserito nell'etichettatura del dispositivo medico insieme alle IFU cartacee. Le eIFU possono essere utilizzate nel caso di dispositivi medici impiantabili, impiantabili attivi, fissi e software (destinati anche ai non addetti ai lavori).
7. Informazioni sugli operatori economici (OE)
L'etichetta contiene solitamente le informazioni relative al produttore. Tuttavia, nel caso di produttori stranieri, le informazioni relative al rappresentante autorizzato devono essere riportate sulle etichette commerciali.
8. Avvertenze e precauzioni
Le avvertenze e le precauzioni devono essere indicate sull'etichetta del dispositivo. Le informazioni su questo aspetto possono essere ridotte al minimo e i relativi dettagli possono essere riportati sull'IFU.
I produttori inoltre tenuti ad adeguarsi ai requisiti di etichettatura specifici del Paese. I requisiti linguistici dipendono dallo Stato membro dell'UE. Ciò può avere un impatto significativo sull'etichettatura, sulle istruzioni per l'uso e sull'imballaggio del dispositivo in termini di tempo e costi.
Questi requisiti aggiuntivi possono ulteriormente appesantire l'onere del produttore con la complessità del processo di etichettatura esistente. Il fallimento dello stesso può diventare molto costoso, comportando il richiamo dei prodotti e le successive fasi di azione correttiva e preventiva (CAPA).
Hai bisogno di assistenza per l'etichettatura secondo il regolamento EU MDR? Freyr servizi completi per l'etichettatura dei dispositivi medici. Contatta subito i nostri esperti in materia di regolamentazione all'indirizzo sales@freyrsolutions.com.









