
3 minuti di lettura
Il 510(k) è una presentazione pre-commercializzazione fatta alla FDA per dimostrare che il dispositivo da commercializzare è altrettanto sicuro ed efficace, cioè sostanzialmente equivalente a un dispositivo legalmente commercializzato (predicato). I dispositivi a rischio moderato sono tenuti a presentare una notifica 510(k), che include una minoranza di dispositivi di Classe I e III e la maggior parte dei dispositivi di Classe II.
Esistono tre (03) tipi di programmi 510(k): tradizionale, abbreviato e speciale. Il percorso per la sicurezza e le prestazioni è stato introdotto nel 2019 e si basa sul programma abbreviato. Il programma eSTAR, introdotto nel 2020, consente di presentare un dispositivo medico completo attraverso un modulo PDF interattivo.
Chi ha bisogno di una certificazione 510(k)?
Il 510(k) è essenzialmente il nome del processo/percorso che i produttori di dispositivi medici che intendono commercializzare i loro dispositivi a rischio moderato-alto negli US intraprendono per dimostrare che il prodotto da commercializzare è sicuro ed efficace quanto un dispositivo legalmente commercializzato.
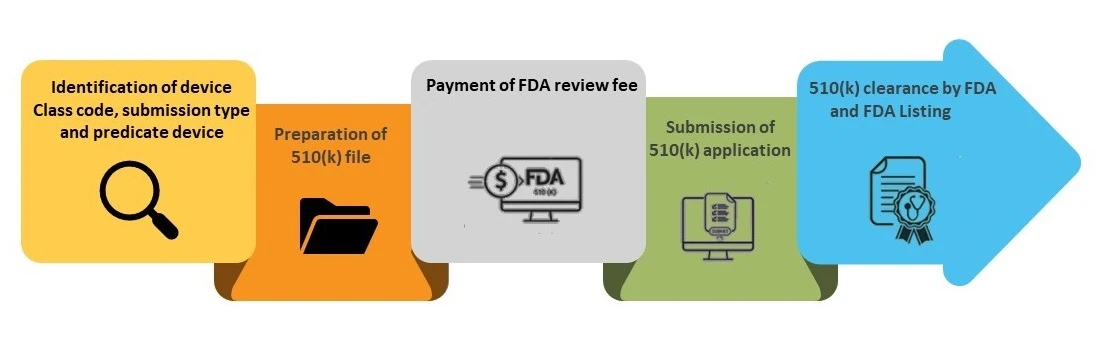
Di seguito viene illustrato il processo di ottenimento dell'autorizzazione 510(k), passo dopo passo.
Fase 1- Identificazione del codice di classe del dispositivo, del tipo di presentazione e del dispositivo predicato
- Identificare il codice prodotto e il numero di regolamentazione - Per determinare i requisiti di test 510(k), è necessario innanzitutto identificare il codice prodotto e il numero di regolamentazione. Si può avviare una ricerca nel database della FDA per trovare il numero di regolamentazione a 7 cifre la cui identificazione corrisponde alla destinazione d'uso del dispositivo in questione.
- Il codice prodotto della FDA è composto da tre lettere. Le informazioni relative alla classificazione del prodotto, alla descrizione della regolamentazione e ai requisiti GMP possono essere trovate utilizzando questo codice.
- Selezione del tipo di presentazione- Un richiedente può scegliere uno dei tre tipi di presentazione menzionati in precedenza. Traditional 510(k) è per le presentazioni iniziali, Special 510(k) è per i produttori di dispositivi medici che desiderano presentare modifiche a un dispositivo esistente e Abbreviated 510(k) può essere scelto quando il dispositivo è conforme agli standard di consenso volontario stabiliti. Nel caso di un Abbreviated 510(k), il richiedente deve fare affidamento sui documenti guida della FDA.
- Identificazione del dispositivo predicato - Il produttore di dispositivi medici deve dimostrare che il dispositivo che intende commercializzare ha la stessa destinazione d'uso e le stesse caratteristiche tecniche del dispositivo legalmente commercializzato, noto anche come dispositivo predicato. In caso di differenze nelle caratteristiche tecniche, il produttore deve dimostrare che la differenza non comporta problemi di sicurezza ed efficacia.
Fase 2- Preparazione del file 510(k)
Il passo successivo è preparare il fascicolo 510(k), le linee guida e le informazioni, disponibili sul sito web della FDA. Include la lista di controllo per l'accettazione per tutti e tre i tipi di programmi 510(k) e un microsito intitolato Contenuti per il 510(k), che include informazioni relative alle dichiarazioni per le indicazioni d'uso, al confronto di equivalenza sostanziale e all'etichettatura proposta, tra le altre informazioni utili.
Fasi del processo di presentazione 510(k)
Fase 3 - Pagamento della tassa di revisione FDA
Tutti i tipi di domande 510(k) sono soggetti alla tassa d'uso. Per l'anno finanziario 2023, la tassa standard per la 510(k) è di 19.870 dollari. Per le aziende certificate dal Centre for Diagnostics and Radiological Health (CDRH), note anche come piccole imprese, la tassa è di 4.967 dollari. La tariffa è soggetta a modifiche nel prossimo esercizio finanziario.
Fase 4 - Presentazione della domanda 510(k)
Il proponente può inviare una copia elettronica (eCopy) o un modello di presentazione e risorse elettroniche (eSTAR) attraverso il portale CDRH.
A partire dal 01 ottobre 2023, tutte le richieste di 510(k), a meno che non siano esentate in base alla guida finale, dovranno essere presentate in formato elettronico utilizzando eSTAR.
Dopo la presentazione del 510(k), viene assegnato un numero di controllo unico, noto come "numero 510(k)" o "numero K". La FDA effettua due controlli di verifica: uno per accertare il pagamento della tassa d'uso corretta e il secondo per verificare se è stata fornita una eCopy o eSTAR valida.
- Entro il giorno 07, la FDA invia una lettera di conferma nel caso in cui la tassa d'uso appropriata sia stata pagata e sia fornita una eCopy o eSTAR valida. In caso contrario, la FDA invia una lettera di sospensione per questioni irrisolte.
- Entro il giorno 15, la FDA effettua una revisione di accettazione e informa il richiedente se il 510(k) è accettato per una revisione sostanziale o se viene posto in stato di rifiuto di accettazione (RTA).
- Entro il giorno 60, la FDA effettua una revisione sostanziale. La FDA comunica tramite
interazione sostanziale per informare che procederà con una revisione interattiva, oppure il 510(k) sarà sospeso e verranno richieste informazioni aggiuntive.
Fase 5 - Autorizzazione FDA e inserimento nel database FDA 510(k)
L'obiettivo della FDA è annunciare la sua decisione sugli Emendamenti alle Tariffe per gli Utenti di Dispositivi Medici (MDUFA) entro 90 giorni FDA. I giorni FDA sono i giorni di calendario tra la data di ricezione del 510(k) e la data di una decisione MDUFA, esclusi i giorni in cui la presentazione è stata sospesa per una richiesta di informazioni aggiuntive. Le decisioni MDUFA per le presentazioni 510(k) includono i risultati di sostanzialmente equivalente (SE) o non sostanzialmente equivalente (NSE).
Quando viene presa una decisione, la FDA invia una lettera di decisione al richiedente via e-mail. Una domanda 510(k) che riceve una lettera di decisione SE è considerata “autorizzata.” Viene quindi elencata nel database 510(k) insieme alle indicazioni per l'uso del dispositivo medico e al riepilogo 510(k) o alla dichiarazione 510(k) come allegati.
Si può concludere che un'attenta pianificazione ed esecuzione, attraverso una documentazione approfondita e una comprensione dettagliata dell'ambiente normativo, sono cruciali per una presentazione 510(k) di successo alla FDA.
Per assistenza con il processo di presentazione 510(k) del vostro dispositivo medico, potete scriverci all'indirizzo sales@freyrsoltions.com, o programmare una chiamata con i nostri esperti, che possono aiutarvi a gestire le procedure. Rimanete informati. Rimanete conformi.









