2 minuti di lettura
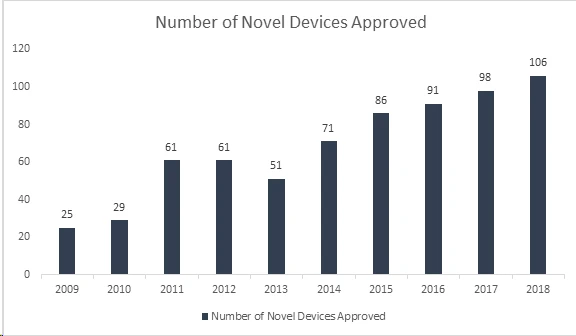
Lo sapevate? La US and Drug Administration (FDA) US ha stabilito un altro record approvando 106 nuovi dispositivi nel 2018, rendendo questo anno il più prolifico in assoluto per i dispositivi medici. Con questo risultato, FDA superato il record quarantennale stabilito nel 2017 con l'approvazione di 99 nuovi dispositivi, dimostrando una crescita continua negli ultimi 8 anni. I dispositivi approvati includono una gamma di prodotti innovativi come sistemi automatizzati di dosaggio dell'insulina per bambini, la valvola cardiaca più piccola al mondo per neonati, la prima app medica mobile per aiutare a gestire i disturbi da abuso di oppioidi e sostanze stupefacenti e tecnologie di intelligenza artificiale che diagnosticano la retinopatia diabetica.
La FDA ha sempre promosso la sicurezza e l'innovazione dei dispositivi medici per garantirne l'alta qualità. Per tenere il passo con il crescente numero di approvazioni di nuovi dispositivi e la loro sicurezza, la FDA sta pianificando di "modernizzare" il percorso di autorizzazione dei dispositivi medici. Secondo l'agenzia, la modernizzazione dell'autorizzazione potrebbe richiedere una nuova autorità. Nel 2018, la FDA e il Center for Devices and Radiological Health (CDRH) hanno pubblicato un documento congiunto in cui si afferma che il 510(k) era uno dei due tipi di presentazione aggiunti alla definizione di dispositivi innovativi. Insieme a questo, anche le esenzioni per dispositivi umanitari (HDE) sono state aggiunte alla definizione di dispositivo innovativo dopo le modifiche apportate al programma per dispositivi medici innovativi del CDRH a seguito del 21° Century Act.
La proposta della FDA e del CDRH è in risposta alla potenziale necessità di perseguire una nuova autorità per modernizzare il processo 510(k). L'obiettivo della proposta è limitare l'uso di dispositivi precedenti, come Sostanzialmente Equivalenti (SE), che hanno più di 10 anni al fine di promuovere l'innovazione. È un passo avanti rispetto alle linee guida rilasciate dall'agenzia nell'aprile 2018. La bozza è stata rilasciata dalla FDA per proporre l'espansione del programma 510(k) abbreviato presso il CDRH della FDA sotto il titolo – “Percorso basato su sicurezza e prestazioni”. È stato introdotto per diminuire l'onere delle disposizioni sui dispositivi medici. L'approccio intende anche aumentare l'efficienza della revisione delle presentazioni 510(k), riducendo così la pressione sull'agenzia.
Ecco alcuni dei punti salienti della guida:
- Il nuovo percorso valuta la sicurezza e l'efficacia dei dispositivi rispetto a standard prefissati di sicurezza e metriche di prestazione.
- Nonostante i nuovi standard, i dispositivi dovranno essere conformi agli standard esistenti per poter essere commercializzati.
- La tecnologia moderna sarà testata rispetto agli standard moderni
- L'approccio promuoverà una maggiore concorrenza per lo sviluppo di dispositivi più sicuri.
Il numero di dispositivi medici che richiedono l'autorizzazione è aumentato esponenzialmente nel corso degli anni. Ciò ha dato ampio margine alla FDA per adottare e implementare misure innovative per migliorare i percorsi di autorizzazione. L'agenzia crede fermamente nei meriti di questa proposta, ma la risposta dell'industria deve ancora essere decifrata.
Con la FDA pubblicare nuovi documenti guida per aggiornare la registrazione dei dispositivi medici, è necessario che produttori di dispositivi medici ne produttori traccia e agiscano di conseguenza. Rimanete aggiornati. Rimanete conformi.









