
3 min read
W dynamicznie zmieniającym się świecie wyroby medyczne zgodność z przepisami nie jest jednorazowym zadaniem — to ciągłe zobowiązanie. Ciągłe monitorowanie i aktualizowanie kluczowych raportów, takich jak raporty z oceny klinicznej (CER), raporty z oceny wydajności (PER) i okresowe raporty dotyczące bezpieczeństwa (PSUR), ma kluczowe znaczenie w całym cyklu życia wyrobu medycznego, od wstępnych badań po nadzór po wprowadzeniu do obrotu. Wraz z ciągłym rozwojem medycyny i zmianami wymagań regulacyjnych, zapewnienie bezpieczeństwa i zgodności wyroby medyczne wyrobów do diagnostyki in vitro poprzez skuteczne tworzenie dokumentacji medycznej pozostaje podstawą sukcesu i długoterminowej rentowności.
Przyjrzyjmy się, w jaki sposób zarządzanie cyklem życia ma zasadnicze znaczenie dla sukcesu urządzenia medycznego
Wprowadzenie na rynek urządzenia medycznego jest kulminacją wieloletnich wysiłków podejmowanych na kilku etapach, takich jak badania, rozwój, próby kliniczne, zgłoszenia do organów regulacyjnych i nadzór po wprowadzeniu na rynek. Proces ten obejmuje wiele lat, a każda faza gromadzi istotne dane w dużych ilościach, które muszą być starannie zestawione i przeanalizowane, aby zapewnić, że urządzenie pozostaje bezpieczne.
Zgodność nie jest jednorazowa, a skuteczne zarządzanie cyklem życia pomaga uniknąć opóźnień, które mogą być kosztowne w późniejszym czasie. Opóźnienia te często wynikają z nieskutecznego planowania nieprzewidzianych zmian regulacyjnych lub niezgodności, które mogły zostać przeoczone. Regularne aktualizacje kluczowych dokumentów, takich jak CER, PER i PSUR, zapewniają, że producenci spełniają zmieniające się wymogi regulacyjne i utrzymują bezpieczeństwo produktów. Zarządzanie cyklem życia zapewnia, że producenci od samego początku planują każdy etap, a tym samym mogą uniknąć wszelkich pułapek i zapewnić pomyślne wejście na rynek i długowieczność swoich urządzeń.
Czy zachowujesz zgodność z przepisami?
Od bandaży po implanty – branża wyrobów medycznych to nowatorski, kreatywny sektor o ogromnych perspektywach. Pomimo silnej chęci producentów do dostarczania na rynek bezpiecznych i wysokiej jakości produktów, nadal istnieje niejasność. Rozporządzenie EU MDR dyrektywę w sprawie wyrobów medycznych (MDD) oraz wyroby medyczne do implantacji (AIMDD). Rozporządzenie to zostało wdrożone w celu wprowadzenia bardziej rygorystycznych kontroli, poprawy bezpieczeństwa pacjentów oraz promowania większej przejrzystości w sektorze opieki zdrowotnej. Istnieje wiele niejasności dotyczących tych norm, a kluczowe kwestie związane z zapewnieniem zgodności są często pomijane. Producenci wyrobów medycznych powinni jednak podjąć kroki w celu uniknięcia trudności regulacyjnych.
Przyjrzyjmy się najczęstszym wyzwaniom stojącym przed producentami urządzeń medycznych
- Prowadzenie CAPA (Corrective and Preventive Action)
- Przestrzeganie procedur składania skarg
- Przestrzeganie procedur czujności
- Ocena kliniczna i nadzór po wprowadzeniu do obrotu
- Łączność z jednostkami notyfikowanymi
Najlepsze praktyki zapewniające zgodność z przepisami
Aby sprostać tym wyzwaniom, we Freyr stworzyliśmy
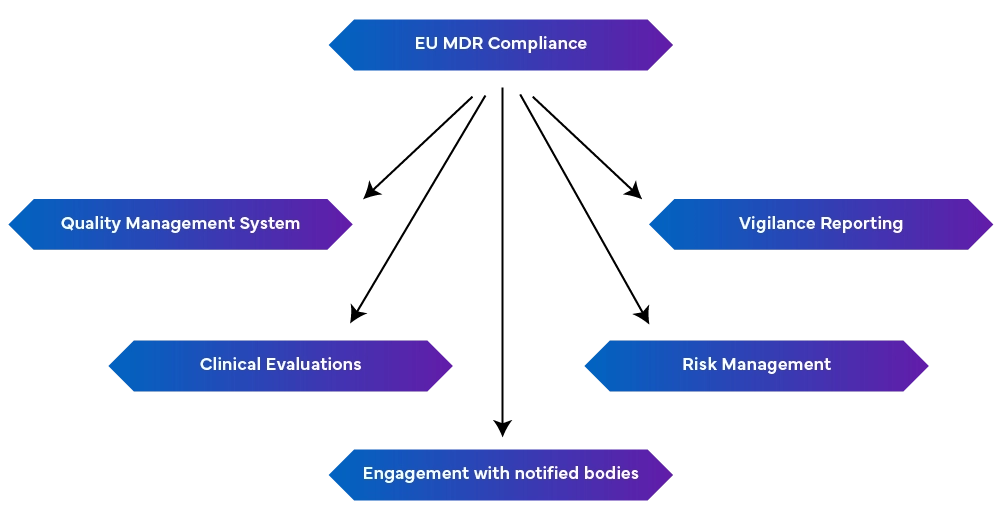
-Solidny system zarządzania jakością (EN ISO 13485:2016): Obejmuje to aspekty produkcyjne, w tym zgodność z przepisami, dokumentację techniczną, deklaracje zgodności UE i zarządzanie ryzykiem.
-Raportowanie czujności: Utrzymanie raportowania czujności jako ciągłego procesu, a nie jednorazowego wysiłku.
- Proaktywne zarządzanie ryzykiem: Identyfikuje i łagodzi ryzyko w całym cyklu życia urządzenia poprzez regularne aktualizacje w celu uwzględnienia pojawiających się zagrożeń.
- Przeprowadzanie ocen klinicznych i nadzoru po wprowadzeniu na rynek: Odbywa się to poprzez wykazanie bezpieczeństwa i wydajności urządzenia przy użyciu danych klinicznych. W razie potrzeby należy przeprowadzić badania kliniczne, a oceny kliniczne powinny być regularnie aktualizowane danymi z nadzoru po wprowadzeniu do obrotu. Wymagane są również okresowe raporty bezpieczeństwa podsumowujące ustalenia.
- Kontakt z jednostkami notyfikowanymi: Zmiany w przepisach mogą wpływać na wymagania MDR, co sprawia, że kluczowe jest utrzymywanie dobrych relacji z jednostkami notyfikowanymi w celu szybkiej aktualizacji i modyfikacji.
Jak ekspert ds. regulacji może pomóc
Próba nadążenia za stale zmieniającym się krajobrazem regulacyjnym może być trudna. Dlaczego więc nie pozwolić ekspertowi poprowadzić Cię przez ten labirynt? Zarządzanie wymaganiami regulacyjnymi w cyklu życia wyrobu medycznego jest nie tylko złożone, ale także wymaga dużych zasobów. W przypadku mniejszych firm, w których wewnętrzne zasoby mogą być lepiej wykorzystane w innych obszarach, pomoc zewnętrznego partnera regulacyjnego może okazać się niezbędna. Oferują oni specjalistyczną wiedzę na temat zmieniających się przepisów i zapewniają, że kluczowe raporty - takie jak CER, PER i PSUR - są stale aktualizowane i zgodne z przepisami.
Można uniknąć kosztownych opóźnień, zapobiec niezgodnościom i usprawnić proces zatwierdzania. Ponadto ekspert ds. regulacji może zaoferować rozwiązania dostosowane do konkretnych potrzeb urządzenia, zapewniając, że nadzór po wprowadzeniu do obrotu, analiza luk i cała dokumentacja zgodności są aktualne. Ich zaangażowanie może uprościć cały proces, zminimalizować ryzyko i zapewnić, że urządzenie spełni oczekiwania w zakresie bezpieczeństwa i wydajności.
Freyr może pomóc we wszystkich potrzebach regulacyjnych dotyczących zarządzania cyklem życia. Reach się us już dziś!









