
Zgodność z przepisami dotyczącymi rejestracji wyrobów medycznych oraz otoczenie regulacyjne w UE
Unia Europejska stanowi jeden z najbardziej uregulowanych i atrakcyjnych pod względem handlowym rynków wyrobów medycznych na świecie. Obejmując 27 member states, 3 EEA krajów oraz Turcję, rynek unijny funkcjonuje w ramach zharmonizowanego systemu regulacyjnego określonego przez EU MDR (2017/745) w odniesieniu do wyroby medyczne oraz unijne rozporządzenie IVDR (2017/746) dotyczące diagnostyki in vitro; region ten zapewnia jednolite standardy bezpieczeństwa i działania we wszystkich kategoriach produktów. Aby legalnie wprowadzić wyrób na rynek UE, producenci muszą uzyskać oznaczenie CE, które potwierdza zgodność z obowiązującymi przepisami w oparciu o klasyfikację ryzyka wyrobu oraz ścieżkę oceny zgodności. Niespełnienie tych wymagań może skutkować poważnymi konsekwencjami, w tym wycofaniem produktu z rynku, zajęciem w urzędzie celnym, zawieszeniem certyfikacji lub całkowitą utratą dostępu do rynku.
Zgodnie z obecnym systemem regulacyjnym:
- Oznaczenie CE jest obowiązkowe dla wszystkich wyrobów medycznych ( wyroby medyczne ) oraz wyrobów do diagnostyki in vitro (IVD) przed wprowadzeniem ich do obrotu.
- Rejestracja wyrobów medycznych w systemie EUDAMED jest wprowadzana etapami, a kilka modułów jest już aktywnych i podlega obowiązkowi stosowania.
- Wyznaczenie europejskiego upoważnionego przedstawiciela (EAR) jest obowiązkowe dla każdego producenta z siedzibą poza UE/EEA/Turcją.
- Bardziej rygorystyczne zasady klasyfikacji określone w rozporządzeniach MDR i IVDR wymagają rzetelnej dokumentacji technicznej, dowodów klinicznych lub dotyczących działania oraz ciągłego monitorowania w całym cyklu życia produktu.
Freyr wspiera producentów na każdym etapie tego procesu, umożliwiając im płynne i zgodne z przepisami wejście na rynek Unii Europejskiej. Od opracowania strategii dotyczącej oznakowania CE i przygotowania dokumentacji technicznej po zarządzanie wnioskami składanymi do jednostek notyfikowanych oraz pełnienie funkcji upoważnionego przedstawiciela w UE — firma Freyr zapewnia pełną zgodność z rozporządzeniami MDR i IVDR oraz płynny dostęp do wszystkich regionów UE member states, EEA oraz Turcji.
Proces zapewnienia zgodności z przepisami UE krok po kroku
Wprowadzenie wyrobu medycznego lub wyrobu do diagnostyki in vitro na rynek UE obejmuje kilka określonych etapów. Oto, w jaki sposób firma Freyr zarządza całym procesem w Państwa imieniu:
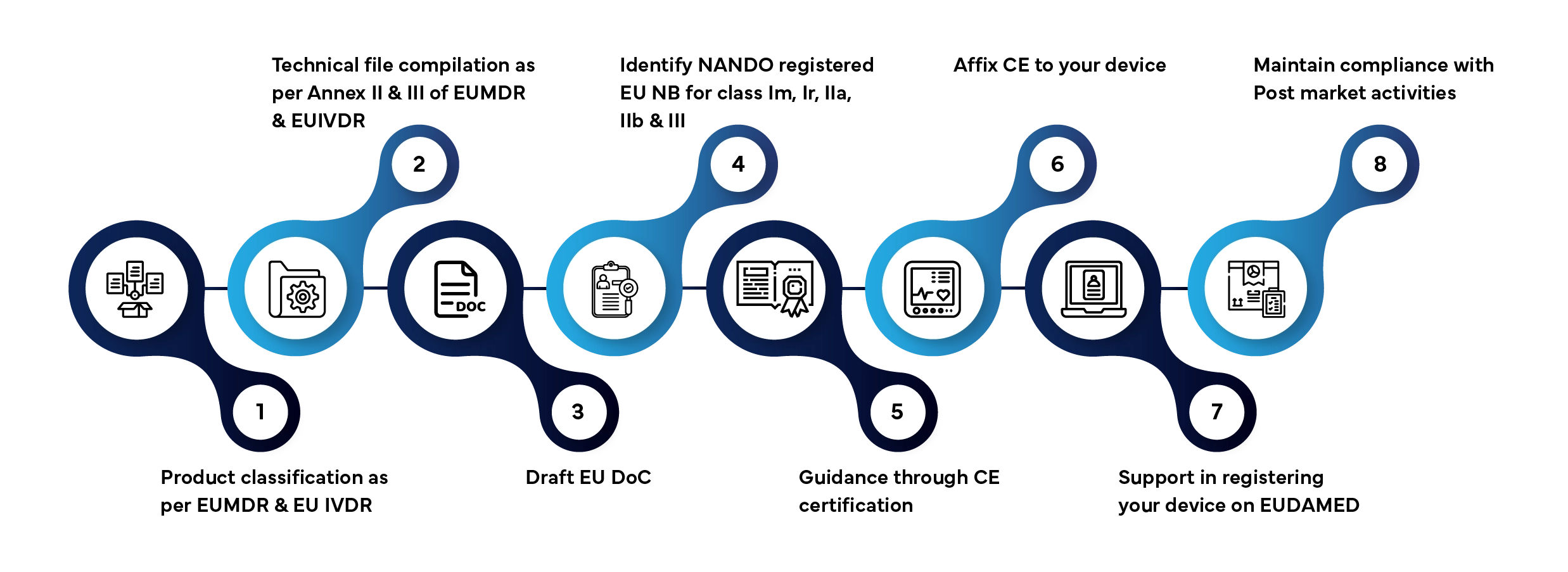
Typowy czas rozpatrzenia wniosku: 3–12 miesięcy, w zależności od klasy urządzenia i kompletności dokumentacji.
Najważniejsze produkty firmy Freyr Medical Device na rynku UE
- EU MDR Strategia regulacyjna i przejście na MDR/IVDR – Opracowujemy plany działania w zakresie regulacji „ end-to-end ”, dostosowane do wymogów rozporządzenia w sprawie wyrobów medycznych ( ) oraz unijnego rozporządzenia w sprawie wyrobów do diagnostyki in vitro (EU IVDR), zapewniając płynne przejście z systemów opartych na dyrektywach oraz zgodność z aktualnymi wymogami UE.
- Dokumentacja techniczna i ocena zgodności – Nasz zespół wspiera Państwa w opracowywaniu, weryfikacji i składaniu dokumentacji technicznej oraz dokumentacji projektowej, pomaga w kontaktach z jednostkami notyfikowanymi, a także zajmuje się oznakowaniem CE, zapewnia wsparcie w zakresie badań bezpieczeństwa i działania wyrobów medycznych oraz ścieżek oceny zgodności.
- Ocena kliniczna/ocena działania: Firma Freyr zapewnia profesjonalne przygotowanie raportów CER i PER, planów badań klinicznych ( PMCF) oraz planów monitorowania po wprowadzeniu do obrotu (PMPF), raportów PSUR, raportów SVR, raportów CPR dotyczących wyrobów do diagnostyki in vitro (IVD), dokumentacji dotyczącej ryzyka oraz treści związanych z oceną biologiczną, gwarantując jasność techniczną i zgodność z przepisami we wszystkich klasach wyrobów.
- Wsparcie w zakresie rejestracji UDI i EUDAMED – Zapewniamy zgodność Państwa systemu unikalnej identyfikacji wyrobów medycznych (UDI) z przepisami oraz pomagamy w rejestracji w europejskiej bazie danych wyrobów medycznych EUDAMED oraz w zarządzaniu cyklem życia wyrobów.
- Europejski upoważniony przedstawiciel (EAR) i reprezentacja lokalna – W przypadku producentów spoza UE/EEA/Turcji pełnimy funkcję Państwa upoważnionego europejskiego przedstawiciela (EAR) i zapewniamy lokalne wsparcie w zakresie zgodności z przepisami w całej UE member states.
- Nadzór po wprowadzeniu do obrotu (PMS) – firma Freyr wspiera Państwa w tworzeniu, wdrażaniu i utrzymywaniu systemów PMS, w tym planu nadzoru po wprowadzeniu do obrotu (PMSP), raport nadzoru po wprowadzeniu do obrotu (PMSR), okresowy raport dotyczący bezpieczeństwa (PSUR), plany działań następczych dotyczących badań klinicznych i działania produktu po wprowadzeniu do obrotu (PMCF/PMPF), zgłaszanie zdarzeń związanych z bezpieczeństwem, działania FSCA, FSN oraz CAPA, zapewniając ciągłe utrzymanie oznaczenia CE oraz trwały dostęp do rynku przy zachowaniu zgodności z przepisami przez cały cykl życia produktu.
- Rejestracja wyrobów medycznych z oznaczeniem CE: Firma Freyr zajmuje się kompleksową obsługą procesu rejestracji w UE w celu uzyskania oznaczenia CE, wspierając ocenę zgodności, przygotowując dokumentację zgodną z wymogami, współpracując z jednostkami notyfikowanymi oraz zapewniając terminowe uzyskanie zatwierdzeń we wszystkich kategoriach wyrobów medycznych i wyrobów do diagnostyki in vitro.
- Wsparcie w zakresie systemów zarządzania jakością: Zapewniamy wsparcie we wdrażaniu i utrzymaniu systemów zarządzania jakością zgodnych z normą ISO 13485, dostosowanych do wymogów dotyczących jakości i bezpieczeństwa określonych w dyrektywie EU MDR/ IVDR oraz oczekiwań jednostek notyfikowanych.
- Zgodność z przepisami dotyczącymi etykietowania: Nasz zespół dba o to, by Państwa etykiety, instrukcje użytkowania, opakowania i symbole były zgodne z wymogami rozporządzeń MDR/IVDR oraz unijnymi wymogami dotyczącymi wielojęzyczności, zapewniając spójność i zgodność z przepisami we wszystkich 27 państwach członkowskich UE member states.
Oferta usług w zakresie pełnomocników upoważnionych przez UE (EAR)

Rejestracja urządzeń w organach UE
W przypadku producentów spoza UE firma Freyr pełni funkcję wyznaczonego upoważnionego przedstawiciela w UE (EAR) zgodnie z wymogami rozporządzeń MDR i IVDR. Jako podmiot z siedzibą w Niemczech firma Freyr wspiera działania związane z rejestracją wyrobów w odpowiednim niemieckim organie właściwym oraz prowadzi wymaganą dokumentację regulacyjną. W przypadku innych podmiotów z UE Member States firma Freyr zapewnia, w stosownych przypadkach, wsparcie regulacyjne, aby pomóc w zapewnieniu zgodności wyrobów z przepisami oraz ułatwić wprowadzenie wyrobu do obrotu w Unii.

Dokumentacja i zapewnienie zgodności
Nasi eksperci ds. regulacji sprawdzają, czy Państwa deklaracja zgodności (DoC), certyfikaty CE oraz dokumentacja techniczna są kompletne, aktualne i zgodne z wymogami rozporządzeń MDR/IVDR. Firma Freyr zapewnia pełną gotowość do przeprowadzenia oceny zgodności oraz złożenia wniosków o oznakowanie CE.

Udzielanie odpowiedzi na zapytania właściwych organów
W razie potrzeby firma Freyr zajmuje się w Państwa imieniu całą bezpośrednią komunikacją oraz odpowiada na prośby o wyjaśnienia kierowane przez właściwe organy UE lub jednostki notyfikowane, zapewniając terminowe i dokładne odpowiedzi oraz pomagając zapobiegać opóźnieniom w procesie uzyskiwania zezwoleń lub przeglądów regulacyjnych po wprowadzeniu produktu do obrotu.

Czujność i informowanie o zdarzeniach
Jako Państwa EAR firma Freyr pełni rolę głównego pośrednika w komunikacji dotyczącej bezpieczeństwa. W razie potrzeby koordynujemy zgłaszanie zdarzeń, działania naprawcze w zakresie bezpieczeństwa w terenie (FSCA) oraz raportowanie w ramach systemu czujności między producentem, pracownikami służby zdrowia i organami regulacyjnymi, aby zapewnić właściwą reakcję i zgodność z przepisami.

Gotowość do kontroli i audytu
Freyr przechowuje całą dokumentację, korespondencję oraz wymagane rejestry na potrzeby audytów i kontroli przeprowadzanych przez organy nadzorcze. Nasz zespół dba o to, by dokumentacja techniczna, etykiety oraz rejestry po wprowadzeniu do obrotu były łatwo dostępne i w pełni zgodne z wymogami rozporządzeń MDR i IVDR.
Dlaczego współpracować z Freyr?
- End-to-end wiedza specjalistyczna w zakresie regulacji, obejmująca cały proces od rejestracji przed wprowadzeniem produktu na rynek po nadzór po wprowadzeniu do obrotu, z uwzględnieniem zarządzania każdym etapem zapewnienia zgodności z przepisami.
- Sprawdzone osiągnięcia obejmujące ponad 2000 pomyślnie przeprowadzonych rejestracji urządzeń z różnych kategorii.
- Silna obecność w UE z siedzibą w Niemczech (EAR), uzupełniona przez lokalnych specjalistów ds. regulacji w Reading oraz wspierana przez globalne zespoły realizacyjne firmy Freyr.
- Indywidualne planowanie transformacji, zapewniające strategiczne wsparcie w celu płynnego i ekonomicznego uzyskania certyfikatu CE.
- Zespoły ds. dostaw obsługujące wiele regionów, korzystające ze wsparcia lokalnego zapewnianego przez oddział firmy Freyr w Niemczech, działający na terenie UE.
- Udokumentowane doświadczenie w zakresie działań naprawczych związanych z rozporządzeniami MDR i IVDR oraz w zakresie dostosowywania starszych wyrobów do nowych wymogów
- Przejrzyste zarządzanie projektem z bezpośrednią komunikacją z jednostką notyfikowaną

Świętujemy Sukcesy Klientów
Wyroby medyczne
Wsparcie w zakresie rejestracji i LR
Globalnie
Freyr okazał się niezastąpionym partnerem w osiąganiu szybkiej globalnej skalowalności dla naszej działalności w zakresie oprogramowania jako wyrobu medycznego (SaMD). Jako startup, zdobycie wiedzy specjalistycznej w zakresie światowych przepisów jest zbyt kosztowne. Konkurencyjne ceny Freyr i dostosowane usługi pozwoliły nam uzyskać tę wiedzę za ułamek kosztów pełnoetatowych zasobów. Szybkość reakcji zespołu i zdolność adaptacji do priorytetów projektu znacznie ułatwiły nasz postęp. Polecamy Freyr każdej firmie poszukującej eksperckiego doradztwa i wsparcia w dziedzinie regulacji wyrobów medycznych.
Arie Henkin
Wiceprezes ds. Jakości i Regulacji, z siedzibą w Australii, wiodąca firma SaMD
Wyroby medyczne
Usługi przedstawicielstwa w Szwajcarii
Japonia i Szwajcaria
Z prawdziwą przyjemnością współpracuję z firmą Freyr i postrzegam ich jako niezwykle cenny zasób oraz rozszerzenie mojego własnego zespołu. Są niezawodni i dokładni, a ich ceny są konkurencyjne. Co więcej, bez wahania ponownie nawiążę współpracę z firmą Freyr.
Darren Mansell
Kierownik ds. Spraw regulacyjnych, z siedzibą w Wielkiej Brytanii, w globalnej firmie zajmującej się projektowaniem i produkcją wyrobów medycznych
Wyroby medyczne
Rejestracja i usługi AR
Malezja i Indonezja
Freyr świadczy niezawodne usługi z ekspertyzą w wielu krajach. Mogę polegać na Freyr, że dostarczy niezbędne informacje do podjęcia świadomej decyzji przed zawarciem formalnej umowy o zakres prac. Gdy projekt jest w toku, zespół Freyr działa profesjonalnie, wykonując pracę z doskonałą komunikacją postępów.







