US FDA Przegląd rejestracji wyrobów medycznych
Stany Zjednoczone Ameryki (USA) są znane jako rynek o wysokim stopniu regulacji dla wyrobów medycznych, z jasno określonymi ścieżkami rejestracji i wymogami. Pierwsze regulacje dotyczące wyrobów medycznych w US pochodzą z 1976 roku i ewoluowały na przestrzeni czasu. Są one regulowane przez Centrum Urządzeń i Zdrowia Radiologicznego (CDRH) pod nadzorem Food and Drug Administration (FDA). Freyr pomógł wielu producentom wyrobów medycznych w spełnieniu wymagań procesu rejestracji wyrobów medycznych US FDA.
![]()
Organ regulacyjny: Agencja Żywności i Leków (FDA)![]()
Regulacja: Tytuł 21 Kodeksu Przepisów Federalnych (21 CFR) Części 800 – 1299![]()
Ścieżka regulacyjna: Zgłoszenie przed wprowadzeniem do obrotu lub Zatwierdzenie przed wprowadzeniem do obrotu lub Klasyfikacja De Novo![]()
Autoryzowany Przedstawiciel: Agent w USA![]()
Wymóg QMS: Rozporządzenie dotyczące Systemu Jakości (QSR) (21 CFR część 820)![]()
Ocena Danych Technicznych: Centrum ds. Wyrobów Medycznych i Zdrowia Radiologicznego![]()
Ważność licencji: Nieograniczone![]()
Wymogi dotyczące oznakowania: 21 CFR Część 801![]()
Format przedłożenia: Papier i płyty CD/DVD![]()
Język: Angielski
Klasyfikacja wyrobów medycznych USA
FDA klasyfikuje wyroby medyczne na 3 kategorie ryzyka: Klasa I, Klasa II i Klasa III. Wyroby klasy I są uznawane za wyroby niskiego ryzyka, natomiast Klasa III wiąże się z wysokim ryzykiem. Wymagania rejestracyjne i ścieżka różnią się w zależności od klasy wyrobu.
| Klasa wyrobu | Ryzyko | Ścieżka rejestracji do zatwierdzenia |
|---|---|---|
| I | Niskie ryzyko | Zwolnienie z 510(k) |
| II | Umiarkowane ryzyko (Z urządzeniem referencyjnym) | Zgłoszenie przed wprowadzeniem do obrotu/510(k) |
Umiarkowane ryzyko (Bez urządzenia referencyjnego) | Wniosek De-Novo | |
| III | Wysokie ryzyko | Zatwierdzenie przedkomercyjne (PMA) |
Agent US FDA
Firmy bez lokalnych biur w US muszą wyznaczyć Agenta FDA w US, aby reprezentował producenta. Agent FDA w US musi mieszkać w US lub utrzymywać miejsce prowadzenia działalności w US. Obowiązki, które ma spełniać agent, są z góry określone przez US FDA w ramach przepisów CFR.
Zapoznaj się z Często zadawanymi pytaniami (FAQ) dotyczącymi Agenta US.
Interaktywne spotkania z US FDA
US FDA wspiera producentów poprzez różnego rodzaju spotkania typu Q-Submission w celu realizacji różnych celów. Takie spotkania z agencją przed rozpoczęciem lub w trakcie opracowywania wyrobu, przed złożeniem wniosków o rejestrację wyrobów medycznych w US FDA, pomagają producentom zoptymalizować harmonogramy i koszty związane z komercjalizacją wyrobu.
Rejestracja wyrobów medycznych USA
Urządzenia mogą być zatwierdzone przez CDRH, FDA za pośrednictwem jednej z różnych ścieżek rejestracji. Są to:
wyroby medyczne klasy I: Wyroby klasy I są zazwyczaj zwolnione z wymogów GMP i zgłoszenia 510(k) i nie wymagają wcześniejszej zgody US FDA na wprowadzenie ich do obrotu w US. Inne wymagania, takie jak rejestracja zakładu, wykaz wyrobów, UDI, PMS itp., muszą być spełnione przez producenta.
wyroby medyczne klasy II: Wyroby średniego ryzyka z zatwierdzonymi wyrobami referencyjnymi 510(k) mogą wybrać Powiadomienie przed wprowadzeniem na rynek 510(k) (PMN), zwane również rejestracją 510(k). Przedmiotowy wyrób powinien wykazać Zasadniczą Równoważność (SE) z zidentyfikowanymi i zgłoszonymi wyrobami referencyjnymi. Jest to najczęściej stosowana ścieżka rejestracji wyrobów w US. Producenci wyrobów średniego ryzyka bez wyrobów referencyjnych mogą ubiegać się o klasyfikację przez US FDA za pośrednictwem wniosków De-Novo.
Wyroby medyczne klasy III: Producenci wyrobów klasy III wysokiego ryzyka muszą złożyć wniosek o Pre-Market Approval (PMA) do US FDA. Wyroby muszą przejść szczegółową ocenę kliniczną, a producent musi przedstawić szczegółowe dane dotyczące bezpieczeństwa i skuteczności z badań klinicznych. US FDA przeprowadzi inspekcję QMS w ramach oceny przed wydaniem Pre-Market Approval dla wyrobu.
Rejestracje wyrobów medycznych spoza CDRH
W zależności od wskazań do stosowania, niektóre produkty graniczne, uznawane w innych krajach za wyroby medyczne, takie jak respiratory chirurgiczne, środki dezynfekujące czy produkty złożone, podlegają nadzorowi innych agencji, np. Centrum Kontroli Chorób (CDC), Krajowego Instytutu Bezpieczeństwa i Higieny Pracy (NIOSH), Agencji Ochrony Środowiska (EPA), Centrum Oceny i Badań Biologicznych (CBER) oraz Centrum Oceny i Badań Leków (CDER).
Wymogi zgodności po zatwierdzeniu dla wyrobów medycznych.
Wszyscy producenci wyrobów muszą przestrzegać poniżej wymienionych wymogów po zatwierdzeniu:
- Wymóg rejestracji i wpisu do wykazu: Placówki wszystkich klas wyrobów muszą być zarejestrowane w bazie danych FURLs, a wyrób musi zostać wpisany do wykazu po uzyskaniu zatwierdzenia i przed wprowadzeniem go na rynek w US. Niektóre wyroby, takie jak wyroby emitujące promieniowanie, muszą spełniać inne wymagania, np. posiadać numer dostępu, zanim zostaną zaimportowane do US.
- Unikalna Identyfikacja Wyrobu: Wszystkie klasy wyrobów muszą spełniać przepisy dotyczące Unikalnej Identyfikacji Wyrobu (UDI), aby wprowadzić je na rynek w US.
- Opłaty rejestracyjne: Producent musi uiszczać roczne opłaty rejestracyjne, aby utrzymać aktywną rejestrację swojego zakładu i móc nadal wprowadzać wyroby medyczne na rynek w US. US FDA stosuje obniżoną strukturę opłat dla mniejszych podmiotów posiadających aktywny Certyfikat Małego Przedsiębiorstwa.
- Audyty jakości: W przypadku wyrobów medycznych, które nie są zwolnione z wymagań GMP, US FDA może w dowolnym momencie przeprowadzić inspekcję zakładu produkcyjnego w celu sprawdzenia zgodności z przepisami dotyczącymi systemów jakości (QSR) zgodnie z 21 CFR 820.
Przebieg procesu
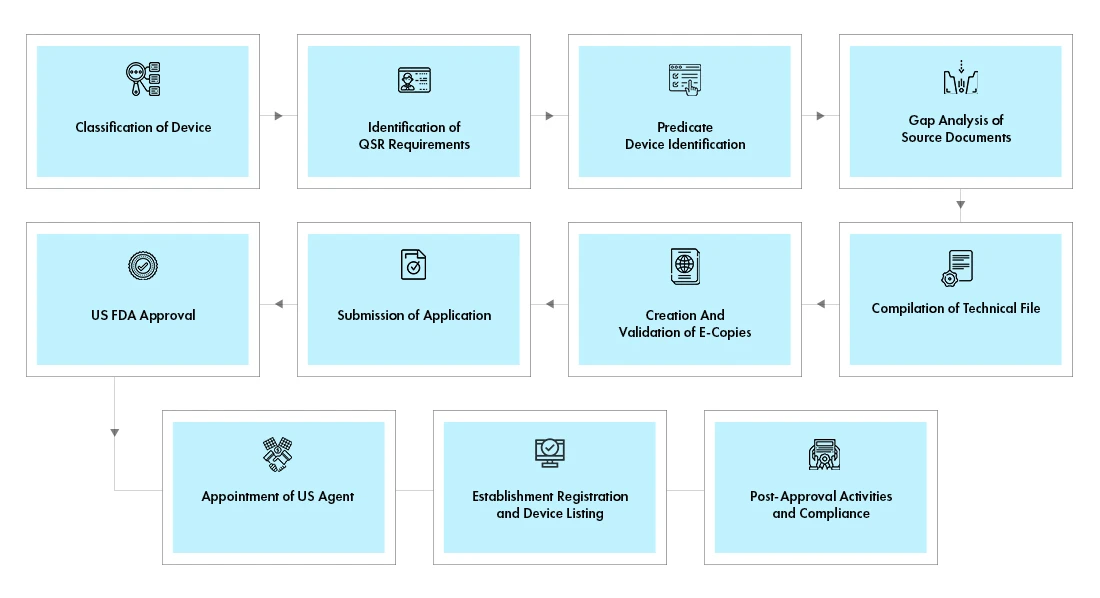
Zarządzanie cyklem życia urządzenia po zatwierdzeniu
Freyr wspiera zagranicznych producentów w zarządzaniu cyklem życia wyrobów medycznych End-to-End, w tym w działaniach po zatwierdzeniu, takich jak:
- Zarządzanie zmianami po zatwierdzeniu – modyfikacje istniejących zatwierdzeń wyrobów medycznych, takie jak dodanie nowych wariantów, akcesoriów; dodanie nowych wskazań do stosowania i inne
- Utrzymywanie zatwierdzeń i rejestracji poprzez terminowe uiszczanie opłat MDUFA do FDA
- Utrzymywanie kontaktu między US FDA a producentem
Freyr posiada wyłączne centrum dostaw w US z profesjonalnym zespołem, aby zapewnić wsparcie regulacyjne producentom w utrzymaniu jakości i bezpieczeństwa niezbędnych do zatwierdzenia. Eksperci Freyr ds. inteligencji bacznie obserwują aktualizacje regulacyjne i informują klientów o krokach, jakie należy podjąć w celu zapewnienia zgodności produktu z obowiązującymi standardami.
Podsumowanie
| Ryzyko | Klasa wyrobu | Audyt QMS | Dostępność urządzenia referencyjnego | Ścieżka regulacyjna | Agent w USA | US FDA Terminy |
|---|---|---|---|---|---|---|
| Niskie ryzyko | I | Nie | N/D | Zwolniony | Tak | 1 miesiąc |
| Średnie ryzyko | II | Tak (po zatwierdzeniu) | Tak | PMN/510(k) | Tak | 9 - 12 Miesięcy |
| Średnie ryzyko | II | Tak (po zatwierdzeniu) | Nie | Wniosek o klasyfikację De-Novo | Tak | 18 - 30 miesięcy |
| Wysokie ryzyko | III | Tak (przed zatwierdzeniem) | N/D | PMA | Tak | 18 - 30 miesięcy |
Usługi Rejestracji Wyrobów Medycznych Freyr
Ekspertyza Freyr
- Należyta staranność regulacyjna
- Dokumentacja urządzeń
- Wsparcie 513(g)
- Rejestracja 510(k)
- Wniosek De-Novo o klasyfikację
- Rejestracja PMA
- Zgodność z 21 CFR 820
- Wsparcie w audycie BIMO
- MDSAP Zgodność
- Wsparcie w zakresie etykietowania
- Wsparcie w publikowaniu i zgłoszeniach
- Agent w USA
- Spotkania dotyczące Q-Submission
- Spotkania RFD i Pre-RFD
- Certyfikacja małych przedsiębiorstw
- Rejestracja zakładu i wykaz wyrobów
- Zgodność regulacyjna dla wyrobów medycznych emitujących promieniowanie
- Zarządzanie zmianami po zatwierdzeniu
- Nadzór pozarynkowy
- Zgodność z UDI
- Doradztwo regulacyjne w zakresie usuwania niedociągnięć.