
Przegląd usług w zakresie zgodności z unijnym rozporządzeniem IVDR
Europejskie Rozporządzenie w sprawie Wyrobów Medycznych do Diagnostyki in vitro (EU IVDR) stanowi nową podstawę regulacyjną dla wprowadzania wyrobów medycznych do diagnostyki in vitro (IVD) na rynek europejski. Rozporządzenie EU IVDR ma zastąpić obecną dyrektywę UE w sprawie wyrobów medycznych do diagnostyki in vitro (EU 98/79/EC). Jako rozporządzenie europejskie, będzie ono obowiązywać we wszystkich Member States UE oraz w państwach Europejskiego Stowarzyszenia Wolnego Handlu (EFTA) bez konieczności transpozycji do prawa poszczególnych państw. Jako sprawdzony partner regulacyjny, Freyr świadczy usługi zgodności z EU IVDR oraz usługi doradcze w zakresie IVDR dla producentów, aby zapewnić zgodność z przepisami dotyczącymi wyrobów medycznych do diagnostyki in vitro i oznakowaniem CE.
Rozporządzenie IVDR – Harmonogram przejściowy
Obowiązujące od 26 maja 2022 r. nowe rozporządzenie UE w sprawie wyrobów medycznych do diagnostyki in vitro (IVDR) jest uważane za znaczącą zmianę w nadzorze regulacyjnym nad wyrobami IVD. Przegląd przejścia na IVDR przedstawiono poniżej –
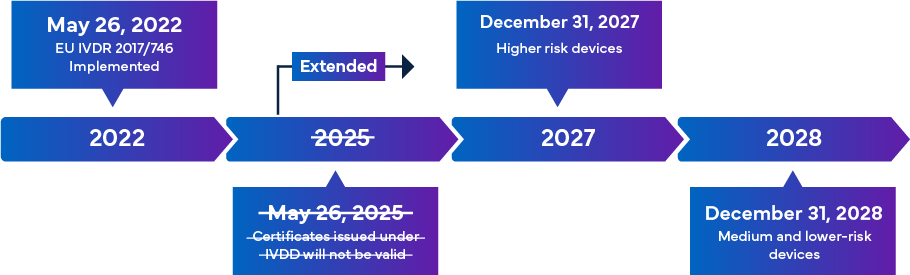
Po pełnym wdrożeniu, zasady zgodności z IVDR zapewniają, że prawie wszystkie wyroby medyczne do diagnostyki in vitro (IVD) wprowadzane na rynek europejski podlegają ocenie jednostki notyfikowanej (NB) IVDR oraz certyfikacji znakiem CE w ramach procesu zatwierdzania IVD. W tym scenariuszu, oprócz dostosowania się do zaktualizowanej klasyfikacji IVDR, istnieje natychmiastowa potrzeba, aby producenci skutecznie przeglądali kluczową dokumentację techniczną w celu pomyślnej rejestracji IVD i uzyskania znaku CE. Wymagania IVDR różnią się w zależności od klasy ryzyka IVD. Ogólnie rzecz biorąc, w celu uzyskania certyfikacji IVD, producenci muszą wykonać:
- Przeglądy dokumentacji technicznej zgodnie z rozporządzeniem IVDR (Rozporządzenie UE 2017/746)
- Przygotowanie raportu z oceny wydajności (PER) dla wszystkich IVD
- Obserwacja działania po wprowadzeniu do obrotu (PMPF) zgodnie z załącznikiem XIII częścią B rozporządzenia IVDR
- Raportowanie w ramach nadzoru zgodnie z artykułem 82 rozporządzenia IVDR
Freyr wspiera klientów w przeprowadzaniu systematycznego przeglądu literatury naukowej oraz pomaga w planowaniu i przygotowaniu PER w celu zapewnienia zgodności z rozporządzeniem w sprawie wyrobów medycznych do diagnostyki in vitro. Freyr posiada dedykowanych ekspertów, którzy świadczą usługi doradcze w zakresie IVDR oraz wsparcie w zakresie nadzoru po wprowadzeniu do obrotu (PMS), które jest integralną częścią systemu zarządzania jakością producenta, wraz z monitorowaniem działania po wprowadzeniu do obrotu (PMPF).
Usługi zgodności z rozporządzeniem IVDR w UE: Ekspertyza i zalety
- Plan przejścia na rzecz zgodności z IVDR
- Przegląd techniczny i analiza luk w zakresie wymogów IVDR dotyczących GSPR (Ogólne Wymagania Bezpieczeństwa i Działania)
- Wsparcie w opracowaniu dokumentacji technicznej zgodnie z wymogami IVDR
- Raporty z oceny ważności naukowej oparte na literaturze i/lub danych wewnętrznych
- Raporty z oceny działania klinicznego oparte na literaturze i/lub danych wewnętrznych
- Dowody kliniczne lub raporty z oceny działania
- Protokoły i raporty z dalszego monitorowania działania po wprowadzeniu do obrotu (PMPF)
- Protokoły i raporty z nadzoru po wprowadzeniu do obrotu (PMS)
- Tworzenie/poprawianie innych dokumentów, takich jak ulotka dołączona do opakowania/IFU (Instrukcje użytkowania), Skrócone Instrukcje Obsługi (QRI), Instrukcja obsługi/użytkownika itp.

- Zapewniona zgodność z rozporządzeniem IVDR, rejestracja IVD i oznakowanie CE
- Głębokie zrozumienie przepisów i doświadczenie w kluczowych obszarach wpływu unijnego rozporządzenia IVDR
- Model dostarczania oparty na silnym zarządzaniu projektami w celu zapewnienia przestrzegania harmonogramu
- Wewnętrzni eksperci NB (przegląd raportu przez interaktywnych recenzentów NB)
- Ukierunkowane zespoły z wiedzą przekrojową w konkretnych obszarach wpływu i kategoriach wyrobów
- Wkład interdyscyplinarny od ekspertów ds. wyrobów medycznych w celu spełnienia wymagań
- Pełny zakres usług w obszarze zgodności, przeglądu i planowania
- Duże doświadczenie w utrzymywaniu spójności w dostarczanych wynikach (czas i jakość)








