Przegląd złożonych wyrobów medycznych
W dynamicznym świecie opieki zdrowotnej i innowacji, produkty złożone z wyrobów medycznych stały się solidnym pomostem łączącym farmaceutyki, wyroby medyczne i produkty biologiczne. Rynek produktów złożonych rozwija się w szybkim tempie, z przewidywaną Złożoną Roczną Stopą Wzrostu (CAGR) na poziomie 8,9% w latach 2023-2030. Sektor produktów złożonych z leków i wyrobów medycznych jest przygotowany na trwały wzrost, wspierany przez postęp technologiczny, ulepszoną infrastrukturę opieki zdrowotnej, płynniejsze ścieżki regulacyjne, strategiczne współprace i zaangażowanie w opiekę skoncentrowaną na pacjencie.
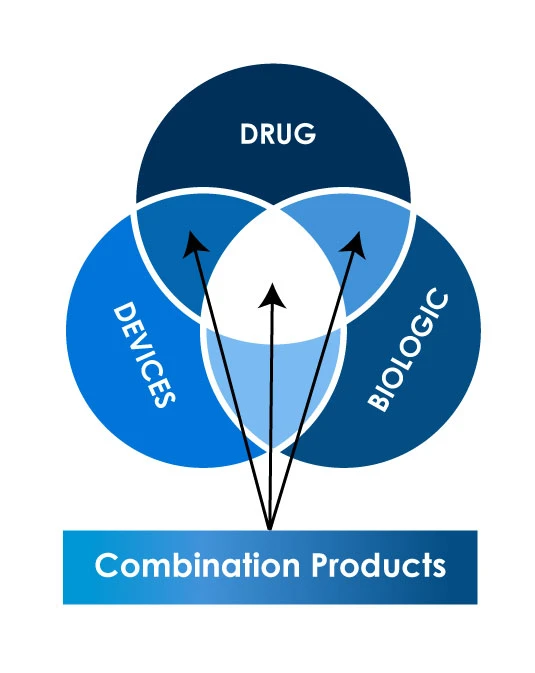
Różne rodzaje produktów złożonych
Globalny scenariusz regulacyjny dla rejestracji produktów złożonych
Interpretacja tego, co stanowi produkt złożony, może różnić się w zależności od kraju, co zwiększa złożoność rejestracji takich produktów w różnych państwach. Ponadto, wymagania regulacyjne i procedury dla produktów złożonych mogą wykazywać różnice w dokumentacji, komunikacji i walidacji. Krajobraz regulacyjny dla rejestracji produktów złożonych może znacznie różnić się na całym świecie. Poniżej przedstawiono kluczowe organy regulacyjne, które globalnie nadzorują te urządzenia.
| Kraj. | Agencja | Główne ośrodki zatwierdzające |
|---|---|---|
| USA | Biuro Produktów Połączonych (OCP) | Centrum Oceny i Badań Leków (CDER) |
| Centrum Oceny i Badań Produktów Biologicznych (CBER) | ||
| Centrum Wyrobów Medycznych i Zdrowia Radiologicznego (CDRH) | ||
| UE | Jednostki Notyfikowane (NB) | Krajowy Organ Kompetentny (Produkty lecznicze) |
| Jednostki Notyfikowane (JN) (wyroby medyczne) | ||
| Japonia | Wydział Oceny i Licencjonowania lub Biuro Produktów Medycznych/Komórkowych i Tkankowych Biura Bezpieczeństwa Farmaceutycznego i Żywności | Dyrektor Wydziału Oceny i Licencjonowania (DMDL), Biuro Bezpieczeństwa Farmaceutycznego i Żywności, Biuro Bezpieczeństwa Farmaceutycznego i Medycznego, Ministerstwo Zdrowia i Opieki Społecznej |
| Chiny | Centrum Administracji ds. Standaryzacji Wyrobów Medycznych (CMDSA) | Centrum Oceny Wyrobów Medycznych (CMDE) |
| Centrum Oceny Leków (CDE) | ||
| Malezja | Krajowa Agencja Regulacji Farmaceutycznych | Krajowa Agencja Regulacji Farmaceutycznych (NPRA) |
| Agencja ds. Wyrobów Medycznych |
Rejestracja produktów złożonych na rynkach międzynarodowych wymaga zindywidualizowanego podejścia, obejmującego ścisłą współpracę z odpowiednimi agencjami zdrowia w celu uzyskania zatwierdzenia. Typowy proces rejestracji produktów złożonych obejmuje następujące etapy:
- Ocena, czy konkretne urządzenie spełnia kryteria klasyfikacji jako produkt złożony.
- Kategoryzowanie wyrobów na podstawie związanych z nimi ryzyk.
- Identyfikacja odpowiednich norm i wymogów dotyczących danych, określonych przez właściwą Agencję Zdrowia.
- Generowanie niezbędnych danych zgodnie z wymogami Agencji.
- Opracowywanie dokumentacji technicznej zgodnie ze szczegółowymi wymogami każdego kraju.
- Złożenie wniosku i odpowiadanie na wszelkie zapytania lub wątpliwości aż do uzyskania zatwierdzenia.
- Zarządzanie cyklem życia wyrobu po zatwierdzeniu.
Nasze Kompetencje
- Wstępna analiza ryzyka.
- Badania rynku – Analizy rynkowe specyficzne dla produktu
- Uzupełnianie kadr
- Opracowanie strategii regulacyjnej
- Potencjalne rynki i drogi
- Dokumentacja projektowa i analiza ryzyka
- System Zarządzania Jakością (QMS) ISO 13485
- Program pojedynczych audytów wyrobów medycznych (MDSAP)
- Ocena wstępna QMS ISO 13485
- Strategia regulacyjna
- Freyr IMPACT (Platforma Informacji Regulacyjnej)
- Weryfikacja i walidacja projektu
- Zarządzanie ryzykiem
- Opracowanie dokumentacji technicznej
- Strategia regulacyjna
- Wymagania regulacyjne
- Narzędzie Freyr rDMS (System Zarządzania Danymi/Dokumentacją)
- Walidacja procesowa i kliniczna
- Ostateczne etykietowanie i Artwork
- Lokalna reprezentacja
- Zgłoszenie regulacyjne.
- Oznakowanie „Conformité Européenne” (CE) Unii Europejskiej (UE) oraz oznakowanie UK Conformity Assessment (UKCA)
- Certyfikacja globalnego dostępu do rynku
- Wsparcie w audycie jednostki notyfikowanej (NB)/jednostki zatwierdzonej
- Lokalna reprezentacja
- Zatwierdzenia regulacyjne
- Nadzór po wprowadzeniu do obrotu (PMS)
- Obserwacja kliniczna po wprowadzeniu do obrotu (PMCF)
- Roczne utrzymanie dokumentacji technicznej (CER/Zarządzanie ryzykiem)
- Odnowienia regulacyjne.
- Wprowadzenia na nowe rynki
- Komunikacja z Właściwym Organem/Jednostką Notyfikowaną/Jednostką Zatwierdzoną
- Zautomatyzowane rozwiązania w zakresie monitorowania bezpieczeństwa stosowania produktów leczniczych (PV)
Dlaczego Freyr?
Rejestracja wyrobów medycznych
- Kompleksowa strategia regulacyjna dla produktu złożonego.
- Wsparcie regulacyjne dla dokumentów rozwoju produktu, takich jak Pliki Historii Projektu (DHFs).
- Strategia zgodności QMS.
- Zgodność regulacyjna, analiza luk i korekta dokumentów technicznych oraz systemów jakości.
- Etykietowanie regulacyjne i usługi pisania technicznego.
- Usługi wywiadu regulacyjnego i rynkowego.
- Usługi tłumaczenia dokumentów i etykiet.
- Współpraca z Agencją Zdrowia i świadczenie usług.
- Usługi regulacyjne dotyczące Artwork.
- Usługi w zakresie monitorowania bezpieczeństwa stosowania produktów leczniczych i PMS.
- Usługi publikowania.
- Usługi pisania tekstów medycznych.

- Pomyślne zgłoszenia dla różnych klas IVD.
- Dedykowany i doświadczony personel do zapewnienia wsparcia regulacyjnego dla wyrobów medycznych i IVD.
- Terminowe złożenie rezultatów.
- Dostęp do lokalnych oddziałów w celu sprostania wyzwaniom Urzędu i specyficznym wymaganiom językowym.
- Wsparcie lokalnego lub prawnego przedstawiciela z wykorzystaniem efektywnego kosztowo modelu.
- Zarządzanie zasobami regulacyjnymi / Usługi zwiększania zasobów kadrowych.