
Przegląd usług zgodności z EU MDR
Unijne Rozporządzenie w sprawie Wyrobów Medycznych (MDR) weszło w życie 26 maja 2021 r., po 3-letnim okresie przejściowym i dodatkowym rocznym przedłużeniu z powodu pandemii COVID-19. Wyroby wprowadzane obecnie na rynek UE muszą być zgodne z tymi przepisami i muszą posiadać certyfikat CE zgodnie z EU MDR, wydany przez jednostki notyfikowane akredytowane na mocy tych przepisów. Wyroby, które już posiadają certyfikat CE zgodnie z EU MDD, mają jednak okresy przejściowe, zanim będą musiały w pełni spełnić wymagania EU MDR. W tym okresie przejściowym wyroby certyfikowane zarówno zgodnie z EU MDD, jak i EU MDR będą współistnieć na rynku na równych zasadach i bez dyskryminacji. Freyr oferuje niezrównane usługi w zakresie zgodności z EU MDR, aby pomóc firmom produkującym wyroby medyczne w terminowym spełnieniu wymagań EU MDR.
Harmonogram przejścia i nowe klasyfikacje wyrobów
Europejskie Rozporządzenie w sprawie Wyrobów Medycznych (MDR) będzie w pełni obowiązywać we wszystkich państwach członkowskich UE oraz państwach Europejskiego Stowarzyszenia Wolnego Handlu (EFTA) od maja 2021 roku i zapewnia producentom czteroletni okres przejściowy na pełną certyfikację EU MDR.
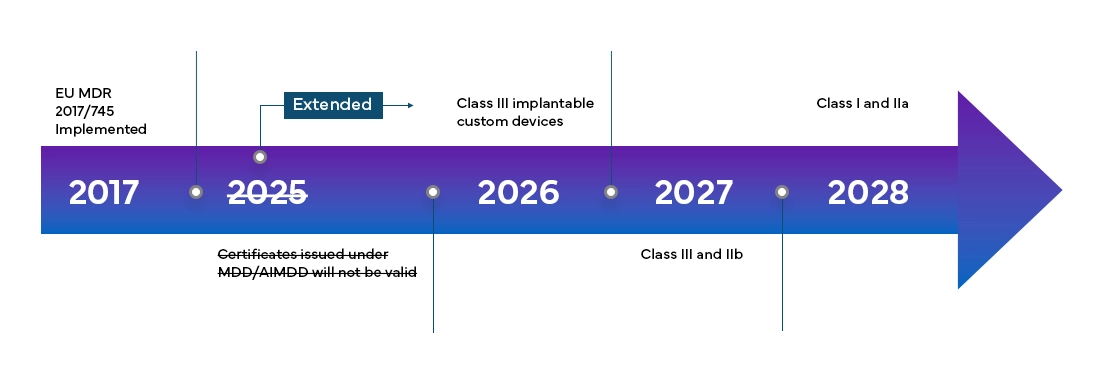
Nowe europejskie rozporządzenie w sprawie wyrobów medycznych (MDR), jak zaobserwowano, wprowadziło również zmiany w istniejącym systemie klasyfikacji wyrobów, takie jak:
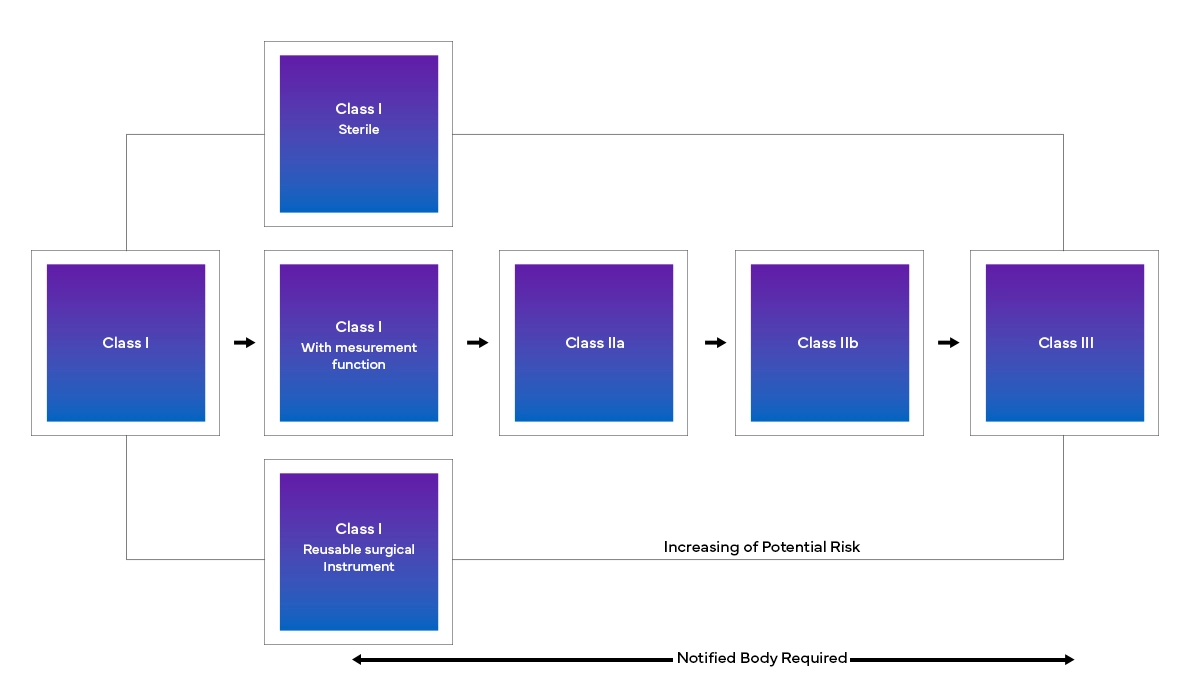
Od identyfikacji dokładnych zmian do ich wdrażania w czasie rzeczywistym, producenci mogą musieć zmierzyć się z szeregiem wyzwań, aby spełnić wymagania EU MDR. Od rozszyfrowania nowej struktury, dokładnej klasyfikacji wyrobu, po zebranie i przesłanie wszystkich danych, producenci będą potrzebować bardziej szczegółowego i interdyscyplinarnego podejścia regulacyjnego, aby sprostać nowym europejskim przepisom dotyczącym wyrobów medycznych. Dzięki rygorystycznej analizie luk, Freyr wspiera klientów w ocenie status quo, a tym samym zapewnia niezbędne działania regulacyjne potrzebne do przejścia i zgodności z EU MDR.
Usługi zgodności z EU MDR
- Opracowywanie jasnej strategii wdrażania Rozporządzenia w sprawie wyrobów medycznych (MDR)
- Zrozumienie nowego prawodawstwa, przeprowadzanie analizy luk w stosunku do obecnych Systemów Zarządzania Jakością (QMS) i obowiązujących procesów
- Opracowywanie szczegółowego planu z podejściem interdyscyplinarnym w celu określenia aspektów systemu jakości, które będą wymagały modyfikacji w związku z nowym unijnym Rozporządzeniem w sprawie wyrobów medycznych
- Tworzenie wielu zespołów do analizy zakresu produktu, klasyfikacji, zarządzania QMS itp. w ramach organizacji, z jednym punktem kontaktowym w każdym zespole
- Alokacja i planowanie zasobów
- Biorąc pod uwagę interakcję Twojego QMS z innymi przepisami i wykorzystując tę okazję do usprawnienia procesów, jednocześnie zapewniając elastyczność w celu uwzględnienia przyszłych zmian.
- Analiza dostępnych danych z testów i sprawdzenie wszelkich dodatkowych wymagań wprowadzonych przez MDR
- Koordynowanie oczekiwań i planu przejścia z Państwa jednostkami notyfikowanymi w UE
- Analiza luk dla istniejących wyrobów medycznych od dyrektywy EU MDD do rozporządzenia EU MDR
- Kompleksowe wsparcie End-to-End w opracowywaniu Raportu Oceny Klinicznej (CER), w tym przegląd literatury zgodnie z wytycznymi europejskiego rozporządzenia w sprawie wyrobów medycznych (EU MDR).
- Usługi End-to-End w zakresie raportów z nadzoru po wprowadzeniu do obrotu (PMSR), okresowych raportów o bezpieczeństwie (PSUR) oraz podsumowania bezpieczeństwa i skuteczności klinicznej (SSCP)
- Wzmocnienie zasobów regulacyjnych z możliwością wdrożenia zasobów lokalnych i zdalnych
- Usługi Europejskiego Upoważnionego Przedstawiciela (EAR)
- Zgodność z MDR i wsparcie w składaniu dokumentacji do Jednostek Notyfikowanych
- Analiza regulacyjna obejmująca proces importu na różnych rynkach regulowanych
- Zgodność QMS i audyty próbne
- System zarządzania dokumentami i narzędzie dla firm objętych MDR
- Klasyfikacja i reklasyfikacja wyrobów według ryzyka
- Wdrożenie UDI i doradztwo
- Usługi nadzoru po wprowadzeniu do obrotu zgodne z Rozporządzeniem UE w sprawie wyrobów medycznych
- Doradztwo w zakresie zarządzania ryzykiem ISO 14971
- Szkolenia wewnętrzne i online
- Osoba odpowiedzialna za usługi i wsparcie w zakresie zgodności regulacyjnej
- Identyfikacja Jednostek Notyfikowanych MDR

W sprawie wsparcia regulacyjnego End-to-End w zakresie EU MDR, skontaktuj się z Freyr.







