

Przegląd rejestracji wyrobów medycznych w Nowej Zelandii
Wyroby medyczne w Nowej Zelandii są regulowane przez Nowozelandzki Urząd ds. Bezpieczeństwa Leków i Wyrobów Medycznych (Medsafe) zgodnie z rozporządzeniami w sprawie leków z 1984 r., ustawą o lekach z 1981 r. oraz rozporządzeniem w sprawie leków (baza danych wyrobów medycznych) z 2003 r. Chociaż zatwierdzenie przed wprowadzeniem na rynek nie jest konieczne, konieczne jest umieszczenie produktów w bazie danych Elektronicznego Systemu Powiadamiania o Wyrobach (WAND) w ciągu 30 dni od wprowadzenia na rynek. Medsafe może zażądać dokumentacji potwierdzającej bezpieczeństwo i skuteczność, takiej jak certyfikacja od uznanych organów, np. jednostki notyfikowanej UE lub Health Canada.
Zespół ekspertów Freyr w dziedzinie regulacji wyrobów medycznych posiada znaczące doświadczenie w prowadzeniu firm produkujących wyroby medyczne przez proces rejestracji Medsafe w Nowej Zelandii.
![]()
Organ regulacyjny: Urząd ds. Bezpieczeństwa Wyrobów Medycznych (Medsafe)![]()
Regulacja:Rozporządzenia (Baza Danych Wyrobów Medycznych) z 2003 roku
Ustawa o Lekach z 1981 roku
Rozporządzenie o Lekach z 1984 roku![]()
Ścieżka regulacyjna: Elektroniczny system powiadamiania o wyrobach wspomagany siecią (WAND)![]()
Autoryzowany Przedstawiciel: Sponsor wyrobu medycznego![]()
Wymóg QMS: Certyfikacja ISO 13485:2016![]()
Ocena Danych Technicznych: Urząd ds. Bezpieczeństwa Wyrobów Medycznych (Medsafe)![]()
Ważność licencji: Wpisy urządzeń w Nowej Zelandii nie wygasają. Urządzenia uznane za stanowiące poważne zagrożenie dla społeczeństwa mogą zostać wycofane z rynku.![]()
Wymogi dotyczące oznakowania: Rozporządzenie 12(4) Przepisów dotyczących Leków z 1984 roku oraz GHTF/SG1/N43:2005![]()
Format przedłożenia: Elektroniczny system powiadamiania o wyrobach wspomagany siecią (WAND)![]()
Język: Angielski
Klasyfikacja wyrobów medycznych w Nowej Zelandii
Wyroby medyczne w Nowej Zelandii są klasyfikowane według ryzyka na klasy I, IIa, IIb, III i AIMD, zgodnie z kryteriami Międzynarodowego Forum Regulatorów Wyrobów Medycznych (IMDRF). Ta klasyfikacja wpływa na zakres niezbędnej kontroli regulacyjnej. Klasyfikacja opiera się na cechach takich jak przeznaczenie wyrobu, czas kontaktu z ciałem, inwazyjność oraz to, czy jest on aktywny, czy nieaktywny. Wyroby wyższej klasy podlegają bardziej rygorystycznemu nadzorowi regulacyjnemu. Medsafe jest organem regulacyjnym w Nowej Zelandii, który nadzoruje te klasyfikacje i przepisy.
| Klasyfikacja wyrobów medycznych Medsafe innych niż IVD | Ryzyko |
|---|---|
| Klasa I Podstawowa | Niskie ryzyko |
| Klasa I pomiarowa | Niskie ryzyko |
| Klasa I sterylna | Niskie ryzyko |
| Klasa IIa | Ryzyko niskie do średniego |
| Klasa IIb | Średnio-wysokie ryzyko |
| Klasa III i Aktywny implantowalny wyrób medyczny (AIMD) | Wysokie ryzyko |
| Klasyfikacja IVD Medsafe | Ryzyko |
|---|---|
| Od lipca 2014 r. Medsafe nie uznaje żadnego systemu klasyfikacji ryzyka dla IVD. Wszystkie IVD zgłoszone do WAND muszą używać kodu klasyfikacji ryzyka IVD. Dyrektor Generalny ds. Zdrowia zezwolił na zwolnienie dla IVD zgodnie z Załącznikiem 1, paragrafem (i) Rozporządzenia w sprawie produktów leczniczych (baza danych wyrobów medycznych) z 2003 r. Jednak dostawcy IVD mogą dobrowolnie zgłaszać swoje wyroby do bazy danych. | |
Upoważniony przedstawiciel/Sponsor wyrobu medycznego
Autoryzowany Przedstawiciel jest nazywany Sponsorem i działa jako pośrednik między producentem a Medsafe. Sponsorzy pełnią funkcję przedstawicieli regulacyjnych dla produktów wprowadzanych na rynek w Nowej Zelandii, składając wnioski WAND i działając jako główny punkt kontaktowy między producentem a Medsafe we wszystkich sprawach związanych z produktem. Dodatkowo, Medsafe pociąga Sponsora do odpowiedzialności za działania w zakresie nadzoru.
Rejestracja wyrobów medycznych w Nowej Zelandii
Rejestracja Wyrobów Medycznych: procedura w Nowej Zelandii oraz procedura wpisu do rejestru WAND w Nowej Zelandii różnią się w zależności od klasy wyrobu.
Urządzenia klasy I – Wymagane jest oświadczenie producenta o zgodności dla niesterylnego, niemierzącego sprzętu klasy I; jednak rzadko jest ono składane do organu regulacyjnego. Zamiast tego, sponsor (lub dostawca) musi wprowadzić szczegóły urządzenia do bazy danych Web Assisted Notification of Devices (WAND) w ramach procesu zgłaszania Medsafe.
Urządzenia innych klas
W Nowej Zelandii sponsorzy lub dostawcy są odpowiedzialni za zapewnienie, że wyroby medyczne spełniają normy takie jak ISO 13485:2016. Bezpośrednie złożenie Deklaracji Zgodności, certyfikacji Systemu Zarządzania Jakością (SZJ) lub dowodów produkcji do Medsafe zazwyczaj nie jest wymagane. Jednak zachowanie tej dokumentacji jest kluczowe dla udowodnienia zgodności na żądanie.
Medsafe priorytetowo traktuje nadzór po wprowadzeniu do obrotu nad szczegółowym zatwierdzeniem przed wprowadzeniem do obrotu wyrobów medycznych. Chociaż audyty nie są rutynowo przeprowadzane w fazie zgłoszenia, Medsafe może je inicjować dla wyrobów o wyższym ryzyku lub po działaniach nadzorczych i zgłoszeniach zdarzeń niepożądanych, zapewniając ciągłe bezpieczeństwo i zgodność.
Po zgłoszeniu urządzenia za pośrednictwem bazy danych WAND, może ono być wprowadzane do obrotu w Nowej Zelandii, pod warunkiem, że dostawca konsekwentnie spełnia przepisy Medsafe. Wymaga to ciągłej zgodności, zwłaszcza z normami monitorowania po wprowadzeniu do obrotu i zgłaszania incydentów. Eksperci ds. wyrobów medycznych w Freyr wspierają usługi związane z poruszaniem się po tych wymaganiach regulacyjnych, zapewniając, że firmy utrzymują zgodność przez cały cykl życia produktu.
Przebieg procesu
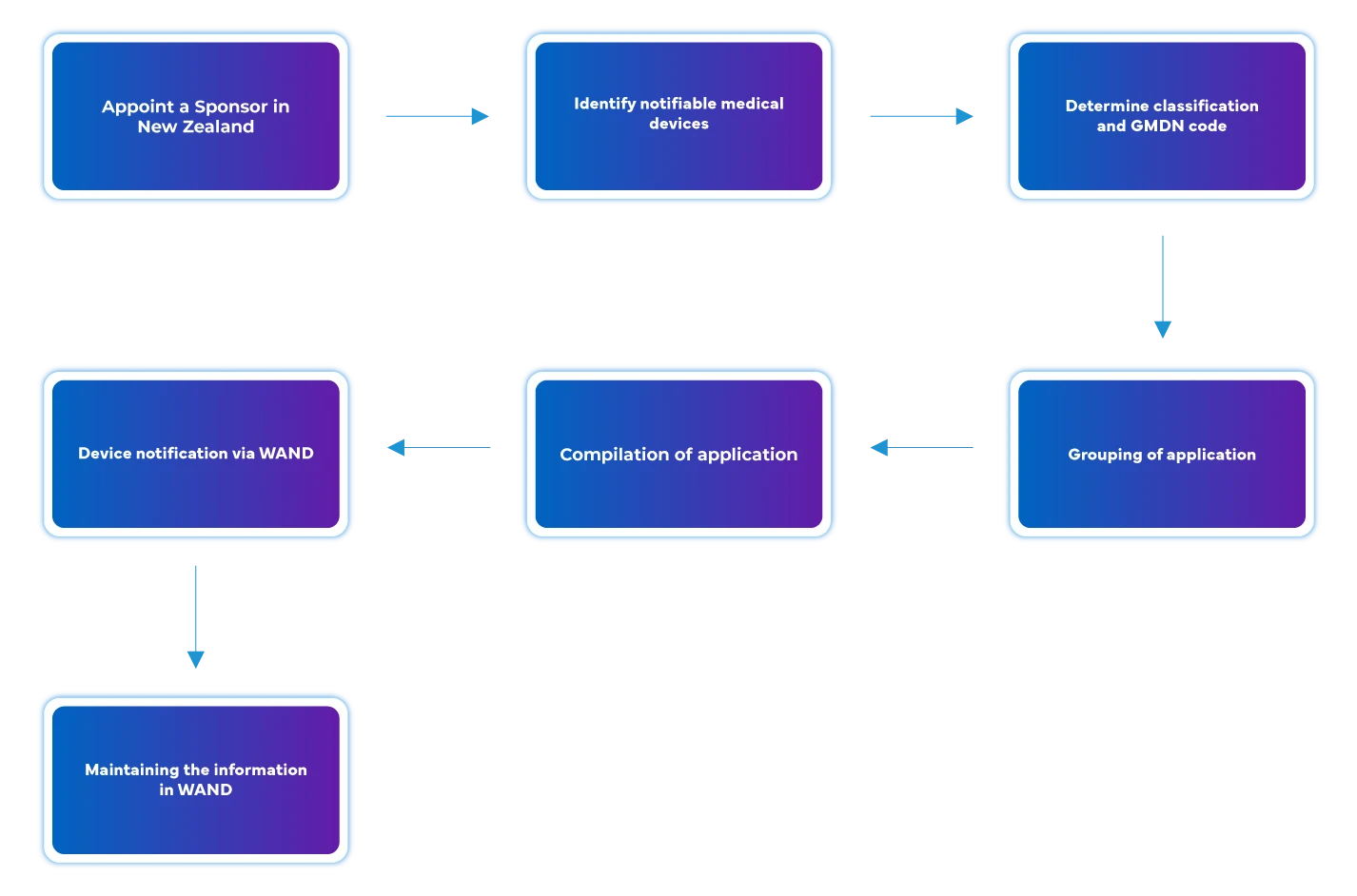
Zarządzanie cyklem życia urządzenia po zatwierdzeniu
Freyr wspiera zagranicznych producentów w zarządzaniu cyklem życia wyrobów medycznych End-to-End, w tym działania po zatwierdzeniu, poprzez powiadamianie władz Nowej Zelandii za pośrednictwem WAND, takie jak –
- Zarządzanie zmianami po zatwierdzeniu – modyfikacje istniejących zatwierdzeń wyrobów medycznych, takie jak dodanie nowych wariantów, akcesoriów; dodanie nowych wskazań do stosowania i inne.
- Utrzymywanie zatwierdzeń i rejestracji.
Dysponując zespołem specjalistów ds. regulacji, Freyr oferuje producentom kompleksowe wsparcie w celu utrzymania standardów jakości i bezpieczeństwa wymaganych do dopuszczenia do obrotu. Specjaliści firmy ds. informacji regulacyjnych skrupulatnie monitorują zmiany w przepisach, zapewniając klientom pełną informację o niezbędnych działaniach w celu utrzymania zgodności ich produktów z obowiązującymi normami.
Podsumowanie
| Ryzyko | Klasa wyrobu | Audyt QMS | Ścieżka regulacyjna | Terminy Medsafe | Ważność rejestracji (lata) |
|---|---|---|---|---|---|
| Niskie ryzyko | Klasa I Podstawowa | Zgodność z ISO 13485:2016 Uwaga- Medsafe nie wymaga audytów QMS, ale zdecydowanie zaleca przestrzeganie ISO 13485:2016 w zakresie jakości i bezpieczeństwa. Medsafe ma prawo do przeprowadzania audytów QMS dla każdej klasy wyrobów, jeśli pojawią się obawy dotyczące bezpieczeństwa lub jakości. | Wykaz WAND (Powiadomienie) | 1 tydzień |
Brak dat ważności |
| Niskie ryzyko | Klasa I pomiarowa | Wykaz WAND (Powiadomienie) | |||
| Niskie ryzyko | Klasa I sterylna | Wykaz WAND (Powiadomienie) | |||
| Ryzyko niskie do średniego | Klasa IIa | Wykaz WAND (Powiadomienie) | |||
| Średnio-wysokie ryzyko | Klasa IIb | Wykaz WAND (Powiadomienie) | |||
| Wysokie ryzyko | Klasa III | Wykaz WAND (Powiadomienie) |
Uwaga: Zgodnie z obowiązującymi przepisami, wpisy urządzeń w Nowej Zelandii nie wygasają, jednak urządzenia, które uznano za stwarzające niedopuszczalne ryzyko dla społeczeństwa, mogą zostać wycofane z rynku. Obecne przepisy mogą jednak zostać zmienione do lat 2026/2027.
Ekspertyza Freyr
- Kompleksowe wsparcie w rejestracji wyrobów medycznych.
- Wsparcie LR
- Wykaz WAND
- Wsparcie w zakresie etykietowania
- Zarządzanie zmianami po zatwierdzeniu
- Przeniesienie licencji
- Usługi składania wniosków i współpracy z WAND