

Przegląd rejestracji urządzeń medycznych w Europie
Unia Europejska, składająca się z 27 member states wyroby medyczne (MDR) 2017/745oraz wyroby medyczne do diagnostyki in vitro (IVDR) 2017/746, które zostały niedawno w pełni wdrożone. Rozporządzenia te stanowią integralną część europejskiego procesu rejestracji wyrobów medycznych i zastąpiły dotychczasowe dyrektywy. Oba rozporządzenia zawierają nowe dodatkowe wymagania, które będą stanowić scentralizowane procedury regulacyjne, których należy przestrzegać przy wprowadzaniu wyroby medyczne dowolnym z 27 krajów. Zagraniczni producenci wyrobów medycznych, którzy nie posiadają fizycznej lokalizacji w Europie, muszą wyznaczyć europejskiego upoważnionego przedstawiciela (EAR), który pomoże im w przestrzeganiu tych przepisów.
![]()
Organ regulacyjny: Właściwy organ krajowy![]()
Rozporządzenie: Przepisy dotyczące wyrobów medycznych (MDR) 2017/745, przepisy dotyczące wyrobów do diagnostyki in vitro 2017/746![]()
Ścieżka regulacyjna: Oznakowanie CE, a następnie rejestracja/zgłoszenie![]()
Upoważniony przedstawiciel: Autoryzowany przedstawiciel europejski (EAR) dla producentów spoza UE![]()
Wymóg SZJ: ISO 13485:2016![]()
Ocena danych technicznych: Jednostka notyfikowana dla oznakowania CE
Klasyfikacja urządzeń
Klasyfikacja urządzenia jest pierwszym krokiem w określeniu ścieżki regulacyjnej dla danego produktu. Istnieje około 22 przepisów wykonawczych dotyczących wyroby medyczne są klasyfikowane jako:
| Klasa | Ryzyko |
|---|---|
| Klasa I | Niski |
| Klasa IIa | Umiarkowany |
| Klasa IIb | Umiarkowany do wysokiego |
| Klasa III | Wysoki |
Podobnie, w przypadku IVD, zaimplementowano około 7 reguł, klasyfikując je do następujących czterech kategorii
| Klasa | Ryzyko |
|---|---|
| Klasa A | Niski |
| Klasa B | Umiarkowany |
| Klasa C | Umiarkowany do wysokiego |
| Klasa D | Wysoki |
Biorąc pod uwagę specjalistyczne instrukcje obowiązujące dla różnych klas, identyfikacja właściwej klasy urządzenia ma kluczowe znaczenie dla określenia ścieżki regulacyjnej.
Autoryzowany przedstawiciel europejski (EAR)
Każdy zagraniczny producent zamierzający wprowadzić swoje urządzenia na rynek w regionie UE jest zobowiązany do wyznaczenia europejskiego autoryzowanego przedstawiciela (EAR ) zgodnie z art. 11 EU MDR i IVDR EU MDR .
Rejestracja urządzeń medycznych
Aby wprowadzić wyroby medyczne do obrotu wyroby medyczne terenie UE, konieczne jest uzyskanie oznakowania CE. Producenci są zobowiązani do identyfikacji i wyznaczenia jednostki notyfikowanej, poddania się ocenie zgodności oraz wydania certyfikatu CE.
Dekodowanie informacji regulacyjnych dotyczących rejestracji urządzenia lub powiadomienia za pośrednictwem internetowego systemu rejestracji może okazać się wyzwaniem bez pomocy eksperta.
Przepływ procesu
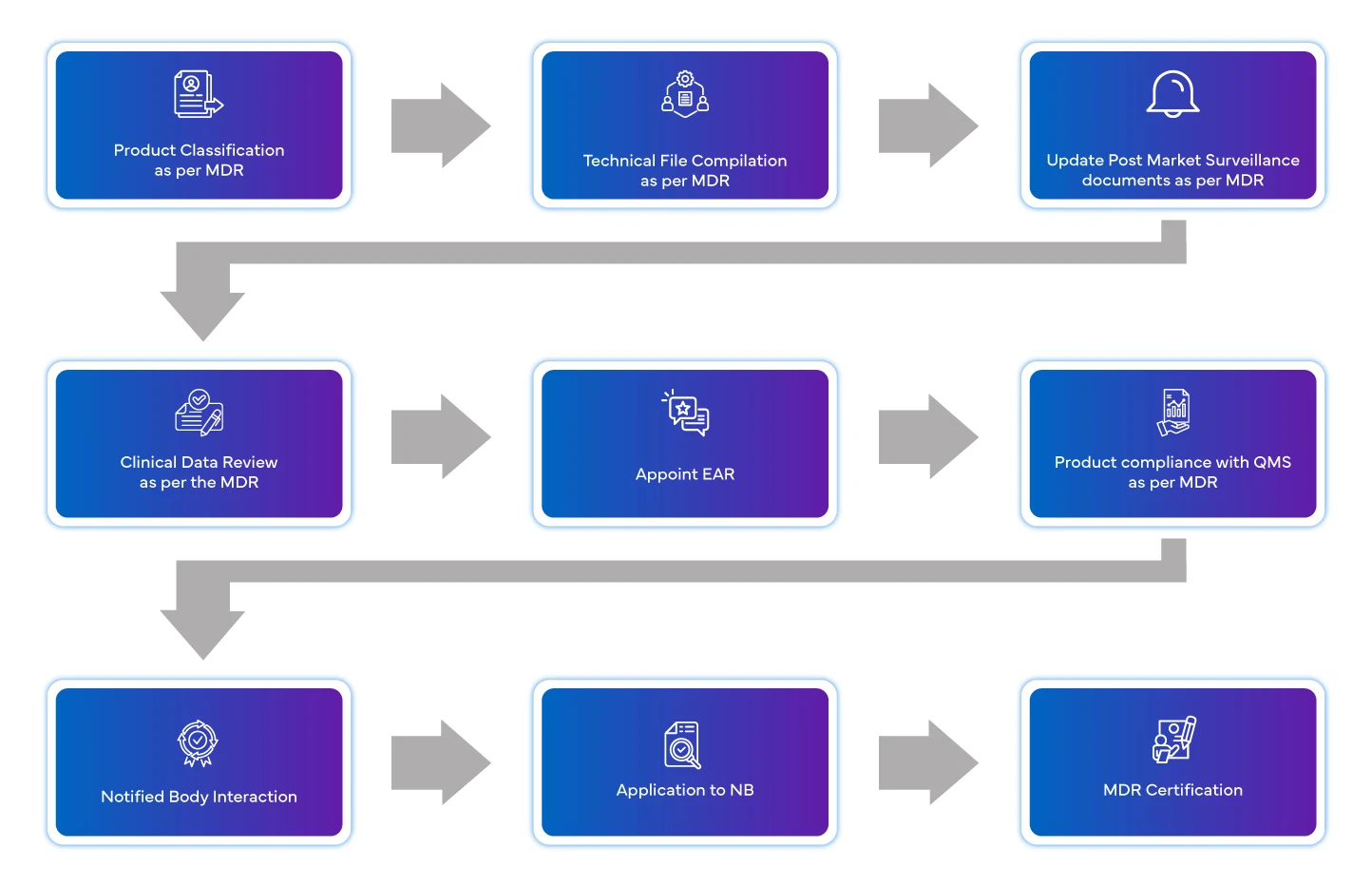
Zarządzanie cyklem życia urządzenia po jego zatwierdzeniu
Europejskie wyroby medyczne kładą obecnie nacisk na znaczeniewymogów dotyczącychdziałań po wprowadzeniu wyrobu do obrotu. Producent ma obowiązek posiadać system zarządzania nadzorem. Należy dostarczać okresowe informacje dotyczące wyrobu.
Freyr może pomóc Państwu w opracowaniu post-market surveillance (PMS) , raportu z nadzoru po wprowadzeniu do obrotu (PMSR), okresowego raportu dotyczącego bezpieczeństwa (PSUR) oraz klinicznej obserwacji po wprowadzeniu do obrotu (PMCF)/obserwacji działania po wprowadzeniu do obrotu (PMPF).
Wsparcie Freyr obejmuje również działania takie jak
- Zarządzanie zmianami po zatwierdzeniu - modyfikacje istniejących zatwierdzeń urządzeń medycznych, takie jak dodanie nowych wariantów, akcesoriów; dodanie nowych wskazań do stosowania, między innymi
- Utrzymanie certyfikatów ISO 13485:2016 i CE
- Odnowienie licencji
- Współpraca między jednostką notyfikowaną a producentem
Rejestracja urządzeń medycznych w Europie
Ekspertyza Freyr
- Europejska klasyfikacja urządzeń medycznych
- Wsparcie europejskiego autoryzowanego przedstawiciela (EAR)
- ISO 14971:2019 Konsultacje w zakresie zarządzania ryzykiem
- Zgodność z normą ISO 13485:2016
- Przegląd, kompilacja i złożenie dokumentacji technicznej/projektowej CE
- Wsparcie EU MDR przejścia EU MDR
- Wsparcie przejściowe UE IVDR
- Clinical Evaluation Reports (CER) wyroby medyczne
- Raporty z oceny działania (PER) dla urządzeń do diagnostyki in vitro
- Powiadomienie/rejestracja wyroby medyczne internetowego systemu rejestracji
- Raport dotyczący strategii regulacyjnej w zakresie wyrobów medycznych
- Wsparcie testowe - biokompatybilność, bezpieczeństwo elektryczne, mechaniczne i wydajnościowe
- Wsparcie w zakresie zgodności etykietowania
- Wsparcie GMP
- Wsparcie w zakresie nadzoru po wprowadzeniu do obrotu
