

Przegląd rejestracji urządzeń medycznych na Tajwanie
Na Tajwanie rośnie popyt na wyroby medyczne. Tajwańska Agencja ds. Żywności i Leków (TFDA) podlegająca Ministerstwu Zdrowia i Opieki Społecznej (MOHW) reguluje wyroby medyczne ustawy o sprawach farmaceutycznych (PAA). Zagraniczni producenci, którzy nie posiadają fizycznego biura na Tajwanie, muszą mieć przedstawiciela na Tajwanie, co jest warunkiem wstępnym rejestracji wyrobów medycznych na Tajwanie.
![]()
Organ regulacyjny: Tajwańska Agencja ds. Żywności i Leków![]()
Rozporządzenie: Ustawa o sprawach farmaceutycznych (PAA) i rozporządzenie w sprawie rejestracji wyroby medyczne![]()
Upoważniony przedstawiciel: Wymagana reprezentacja agenta na Tajwanie![]()
Wymóg SZJ: Dokumentacja systemu jakości (QSD) ISO 13485![]()
Ocena danych technicznycha: Dział wyroby medyczne kosmetyków![]()
Ważność licencji: QSD - 3 lata; Rejestracja produktu - 5 lat![]()
Wymagania dotyczące etykietowania: Artykuł 75 ustawy o produktach farmaceutycznych![]()
Format zgłoszenia: Papier![]()
Język: Angielski i chiński
Tajwańska klasyfikacja urządzeń medycznych
TFDA klasyfikuje wyroby medyczne 3 klas w oparciu o ryzyko: klasa I dla wyrobów niskiego ryzyka, klasa II dla wyrobów umiarkowanego ryzyka i klasa III dla wyrobów wysokiego ryzyka. Konieczność posiadania wyrobu odniesienia stanowi wyzwanie dla wprowadzania nowych wyrobów na rynek. Dodatkową komplikacją jest wydłużenie czasu procedury dla wyrobów klasy II i III, które wymagają dokumentacji systemu jakości. Wszystkie importowane wyroby medyczne uzyskać certyfikat rejestracji od TFDA.
| Klasa urządzenia | Ryzyko |
|---|---|
| Klasa I | Niskie ryzyko |
| Klasa II | Umiarkowane ryzyko |
| Klasa III | Wysokie ryzyko |
Reprezentacja agenta na Tajwanie
Zagraniczni producenci nie posiadający fizycznego biura na Tajwanie powinni wyznaczyć tajwańskiego agenta jako warunek wstępny wprowadzenia urządzeń na rynek tajwański. Wyznaczenie organizacji zewnętrznej jako tajwańskiego agenta zamiast dystrybutora zapewnia elastyczność w zakresie poszukiwania wielu dystrybutorów w celu lepszej penetracji rynku. Tajwański agent musi posiadać osobowość prawną z siedzibą na Tajwanie, certyfikowaną licencją na sprzedaż produktów farmaceutycznych.
Rejestracja urządzeń medycznych na Tajwanie
Zanim wyrób medyczny będzie mógł być sprzedawany na Tajwanie, oprócz rejestracji wyrobu medycznego wymagana jest rejestracja dokumentacji systemu jakości (QSD) dla zakładu produkcyjnego. Rejestracja QSD nie jest wymagana tylko w przypadku wyroby medyczne klasy I (niejałowych). Licencja QSD (otrzymywana po zatwierdzeniu rejestracji QSD) na Tajwanie jest podobna do dobrej praktyki wytwarzania (GMP) dla wyroby medyczne.
TFDA ogłosiła, że od 1 czerwca 2022 r. posiadacze licencji na wyroby medyczne klasy III wyroby medyczne zobowiązani do wprowadzenia identyfikatorów UDI i odpowiednich informacji o produktach do bazy danych UDI (UDID). Producenci wyrobów medycznych będą również zobowiązani do umieszczania identyfikatorów UDI na etykietach produktów. Ponadto od 1 czerwca 2023 r. wyroby medyczne klasy II wyroby medyczne musiały spełniać odpowiednie przepisy dotyczące identyfikatorów UDI.
Przepływ procesu
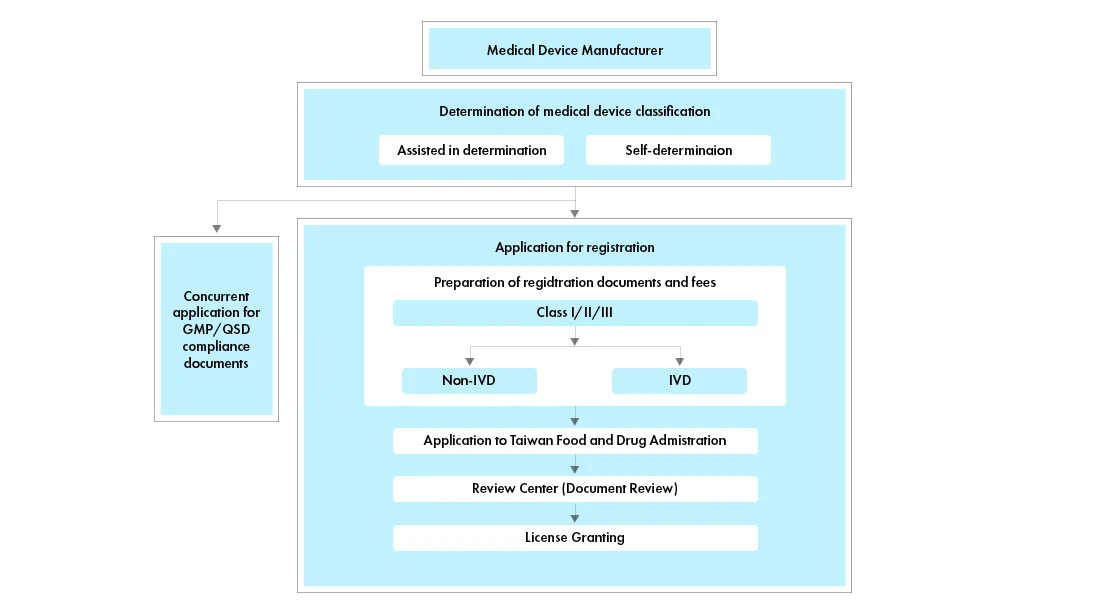
Zarządzanie cyklem życia wyrobów medycznych po ich zatwierdzeniu
Freyr wspiera zagranicznych producentów w end-to-end zarządzaniu cyklem życia urządzeń end-to-end , w tym w działaniach po uzyskaniu zatwierdzenia, takich jak:
- Zarządzanie zmianami po zatwierdzeniu - modyfikacje istniejących zatwierdzeń urządzeń medycznych, takie jak dodanie nowych wariantów, akcesoriów; dodanie nowych wskazań do stosowania, między innymi
- Utrzymanie zatwierdzeń i rejestracji poprzez terminowe uiszczanie opłat administracyjnych i rejestracyjnych.
- Odnowienie licencji
- Współpraca między TFDA a producentem
- Zarządzanie importem
Freyr specjalizuje się w obsłudze potrzeb regulacyjnych wyroby medyczne Tajwanie. Dzięki rozległej sieci kontaktów Freyr pomaga w wyznaczeniu niezawodnego lokalnego przedstawiciela, którego obecność ma ogromne znaczenie podczas Nadzór po wprowadzeniu do obrotu. Nasi eksperci pomagają również w wyborze odpowiedniego wyrobu referencyjnego i istniejących zatwierdzeń z innych rynków, aby wesprzeć wejście nowego wyrobu na rynek.
Podsumowanie
| Klasa urządzenia | Kryteria ryzyka / klasyfikacji | QMS | Rejestracja produktu |
|---|---|---|---|
| Klasa I | Niskie ryzyko | Zwolnione (niesterylne urządzenia klasy I) | Tak |
| Klasa II | Umiarkowane ryzyko | QSD | Tak |
| Klasa III | Wysokie ryzyko | QSD | Tak |
Ekspertyza Freyr
- Należyta staranność regulacyjna
- Oficjalna klasyfikacja
- Zatwierdzenia QSD
- Rejestracja urządzenia
- Przedstawiciel prawny
- Obsługa etykietowania
- Wsparcie w zakresie tłumaczeń
- Identyfikacja i kwalifikacja dystrybutorów
- Nadzór po wprowadzeniu na rynek
- Zarządzanie zmianami po zatwierdzeniu
- Odnowienie i przeniesienie licencji
- Składanie wniosków i współpraca
