

Przegląd rejestracji urządzeń medycznych w Turcji
W ciągu ostatniej dekady turecki rynek wyrobów medycznych odnotował znaczny i stały wzrost. Od 2021 r. rejestracja wyrobów medycznych w Turcji wymaga zgodności z unijnymwyroby medyczne (MDR) 2017/745oraz wyroby medyczne do diagnostyki in vitro (IVDR) 2017/746. Ta poprawa warunków handlu międzynarodowego skłoniła kilka globalnych firm do wprowadzenia swoich wyroby medyczne rynek turecki.
![]()
Organ regulacyjny: Turecka wyroby medyczne ds. Leków i wyroby medyczne (TITCK)![]()
Rozporządzenie: Przepisy dotyczące wyrobów medycznych (MDR) 2017/745, przepisy dotyczące wyrobów do diagnostyki in vitro 2017/746![]()
Ścieżka regulacyjna: Oznakowanie CE jest obowiązkowe, po czym następuje rejestracja/zgłoszenie w systemie śledzenia produktów (UTS).![]()
Lokalny autoryzowany przedstawiciel w Turcji: Autoryzowany przedstawiciel europejski (EAR) dla producentów zagranicznych (spoza UE / spoza Turcji)![]()
Wymóg SZJ: ISO 13485:2016![]()
Ocena danych technicznych: Jednostka notyfikowana dla oznakowania CE![]()
Ważność licencji: Bez ograniczeń![]()
Format zgłoszenia: Papier![]()
Tłumaczenie: Przetłumaczone dokumenty w języku tureckim
Klasyfikacja urządzeń
Turcja stosuje taką samą klasyfikację urządzeń medycznych, jak podano w EU MDR i IVDR. Określenie klasyfikacji urządzenia może być wyzwaniem, dlatego też wsparcie doświadczonego konsultanta ds. regulacji prawnych ma tutaj kluczowe znaczenie.
Klasy urządzeń medycznych -
| Klasa | Ryzyko |
|---|---|
| Klasa I | Niski |
| Klasa IIa | Umiarkowany |
| Klasa IIb | Umiarkowany do wysokiego |
| Klasa III | Wysoki |
Klasy urządzeń do diagnostyki in vitro -
| Klasa | Ryzyko |
|---|---|
| Klasa A | Niski |
| Klasa B | Umiarkowany |
| Klasa C | Umiarkowany do wysokiego |
| Klasa D | Wysoki |
Lokalny autoryzowany przedstawiciel w Turcji
Obecnie, dzięki umowie o unii celnej, producenci z UE nie muszą wyznaczać lokalnego autoryzowanego przedstawiciela w celu wprowadzenia swoich urządzeń na rynek.
Inni zagraniczni producenci są zobowiązani do wyznaczenia europejskiego autoryzowanego przedstawiciela (EAR ) w celu wprowadzenia urządzeń na rynek turecki.
Rejestracja urządzeń medycznych
Oznakowanie CE to zgodność wymagana przez producentów w celu wprowadzenia ich urządzeń na rynek turecki. Oznakowanie CE jest wydawane w drodze oceny zgodności przeprowadzanej przez jednostkę notyfikowaną. Obecnie Turcja jest upoważniona do wyznaczania jednostek notyfikowanych zgodnie z EU MDR i IVDR.
Firmy są zobowiązane do zarejestrowania się w Centralnym Systemie Rejestracji (MERSIS) i zarejestrowania urządzenia w Systemie Śledzenia Produktów (UTS).
Przepływ procesu
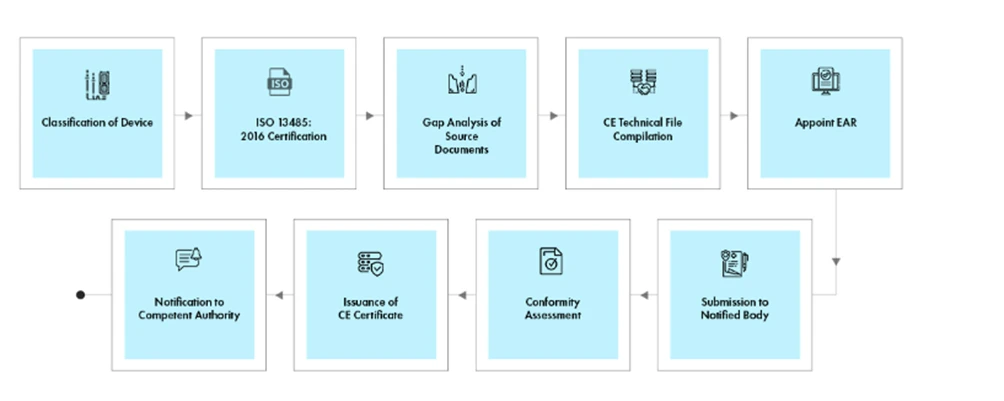
Zarządzanie cyklem życia urządzenia po jego zatwierdzeniu
Freyr zagranicznych producentów w end-to-end zarządzaniu cyklem życia wyrobów end-to-end , w tym w działaniach po uzyskaniu zatwierdzenia, takich jak:
- Zarządzanie zmianami po zatwierdzeniu - modyfikacje istniejących zatwierdzeń urządzeń medycznych, takie jak dodanie nowych wariantów, akcesoriów; dodanie nowych wskazań do stosowania, między innymi
- Utrzymanie certyfikatów ISO 13485:2016 i CE
- Odnowienie licencji
- Współpraca między jednostką notyfikowaną a producentem
Ze względu na zaangażowanie różnych organów certyfikujących zagraniczni producenci muszą przestrzegać wielu zestawów przepisów w każdym indywidualnym procesie zatwierdzania wyrobów. Uzyskanie oznakowania CE i dalsze przestrzeganie przepisów stanowych wymaga rozległej wiedzy regulacyjnej. Czasami, bez sprawdzonego partnera regulacyjnego, poruszanie się po wszystkich wymaganiach dotyczących wyrobów może być trudne dla podmiotów wchodzących na rynek. Aby pomóc producentom, Freyr end-to-end usługi end-to-end w celu przyspieszenia procesu zatwierdzania wyroby medyczne.
Ekspertyza Freyr
- Europejska klasyfikacja urządzeń medycznych
- Wsparcie europejskiego autoryzowanego przedstawiciela (EAR)
- Rejestracja urządzenia i powiadomienie o produkcie w Turcji
- ISO 14971:2019 Konsultacje w zakresie zarządzania ryzykiem
- Zgodność z normą ISO 13485:2016
- Przegląd, kompilacja i złożenie dokumentacji technicznej/projektowej CE
- Wsparcie EU MDR przejścia EU MDR
- Wsparcie przejściowe UE IVDR
- Clinical Evaluation Reports (CER) wyroby medyczne
- Raporty z oceny działania (PER) dla urządzeń do diagnostyki in vitro
- Powiadomienie/rejestracja wyroby medyczne internetowego systemu rejestracji
- Raport dotyczący strategii regulacyjnej w zakresie wyrobów medycznych
- Wsparcie testowe - biokompatybilność, bezpieczeństwo elektryczne, mechaniczne i wydajnościowe
- Wsparcie w zakresie zgodności etykietowania
- Wsparcie GMP
- Wsparcie w zakresie nadzoru po wprowadzeniu do obrotu
