
Przegląd rejestracji wyrobów medycznych w Egipcie
Od września 2018 roku rejestracja wyrobów medycznych stała się obowiązkowa w Egipcie. Przemysł wyrobów medycznych w tym kraju odnotował stały wzrost, co czyni go atrakcyjnym rynkiem dla producentów i dystrybutorów. Z wyceną 4,0 miliarda dolarów w 2021 roku, egipski rynek wyrobów medycznych ma osiągnąć skumulowany roczny wskaźnik wzrostu (CAGR) powyżej 3% w latach 2022-2027. Import w dużej mierze zaspokaja zapotrzebowanie na wyroby medyczne w Egipcie, biorąc pod uwagę stosunkowo niską produkcję lokalną. Co ważne, egipski rynek wyrobów medycznych jest drugim co do wielkości w regionie Bliskiego Wschodu i Afryki Północnej (MENA). Ten przegląd bada kluczowe aspekty egipskiego procesu rejestracji, oferując wgląd w ramy regulacyjne i wymagania dotyczące wprowadzania innowacyjnych wyrobów medycznych na czoło egipskiego sektora opieki zdrowotnej.
Organ regulacyjny: Egipska Agencja Leków (EDA)
Regulacja: Egipska Ustawa o Wyrobach Medycznych Prawo nr 10 z 2003 r.
Ścieżka regulacyjna: Rejestracja produktu (tryb normalny i przyspieszony) oraz oficjalna klasyfikacja
Lokalny Autoryzowany Przedstawiciel w Egipcie: Egipski Posiadacz Rejestracji (ERH)
Wymóg QMS: ISO 13485
Ocena danych technicznych: Centrum Polityki i Planowania Leków (DPPC) oraz Centralna Administracja ds. Farmaceutycznych (CAPA).
Ważność licencji: Dziesięć (10) lat
Format składania: Papierowy i elektroniczny
Tłumaczenie: Przetłumaczone dokumenty w języku arabskim i angielskim
Klasyfikacja wyrobów
W Egipcie klasyfikacja wyrobów medycznych jest zgodna z europejskim systemem klasyfikacji, który kategoryzuje wyroby medyczne na podstawie ich przeznaczenia i potencjalnego ryzyka związanego z ich użyciem. Producenci powinni określić prawidłową klasyfikację swoich wyrobów, aby zapewnić zgodność z wymaganiami regulacyjnymi i uzyskać niezbędne zezwolenia na wprowadzanie do obrotu i dystrybucję w Egipcie.
Klasy wyrobów medycznych
| Klasa | Ryzyko |
|---|---|
| Klasa I | Niski |
| Klasa IIa | Nisko-średni |
| Klasa IIb | Średnio-wysoki |
| Klasa III | Wysoki |
Lokalny Autoryzowany Przedstawiciel w Egipcie
Firmy produkujące wyroby medyczne z siedzibą poza Egiptem muszą wyznaczyć lokalnego agenta, zwanego „Egipskim Posiadaczem Rejestracji (ERH)”, który będzie w ich imieniu zajmował się składaniem wniosków rejestracyjnych i dossier do EDA. ERH pełni funkcję łącznika między producentem a organem regulacyjnym, zapewniając dokładne przygotowanie i złożenie całej wymaganej dokumentacji oraz weryfikując, czy wyrób medyczny spełnia standardy bezpieczeństwa, jakości i skuteczności EDA. Ponadto ERH jest odpowiedzialny za przechowywanie dokumentacji rejestracyjnej, zgłaszanie incydentów lub wycofań oraz zapewnienie ciągłego przestrzegania wszystkich obowiązujących norm i przepisów przez cały cykl życia urządzenia. Egipski Posiadacz Rejestracji (ERH) jest w pełni odpowiedzialny za zapewnienie rejestracji wyrobu medycznego w EDA, w szczególności w ramach Centralnej Administracji Wyrobów Medycznych. Rola ta obejmuje zapewnienie zgodności urządzenia z wymogami regulacyjnymi EDA dotyczącymi wprowadzania do obrotu i dystrybucji w Egipcie.
Rejestracja wyrobów medycznych
Uzyskanie Pozwolenia na dopuszczenie do obrotu produktu leczniczego dla wyrobu medycznego w Egipcie obejmuje kilka etapów, w tym przygotowanie wymaganej dokumentacji, złożenie wniosku do EDA, przestrzeganie wymagań dotyczących klasyfikacji i systemu jakości, powołanie ERH w razie potrzeby oraz spełnienie obowiązków po wprowadzeniu na rynek. Proces rejestracji jest kluczowy, aby zagwarantować, że wyroby medyczne są zgodne z ustalonymi normami bezpieczeństwa, jakości i skuteczności, określonymi przez Egipski Organ Regulacyjny. Wymagana dokumentacja może się różnić w zależności od wybranej ścieżki rejestracji, ale zazwyczaj obejmuje następujące elementy:
- Certyfikat CE (jeśli dotyczy).
- Certyfikat Wolnej Sprzedaży (CFS).
- Certyfikacja ISO 13485.
- Deklaracja Zgodności (DOC).
Przebieg procesu
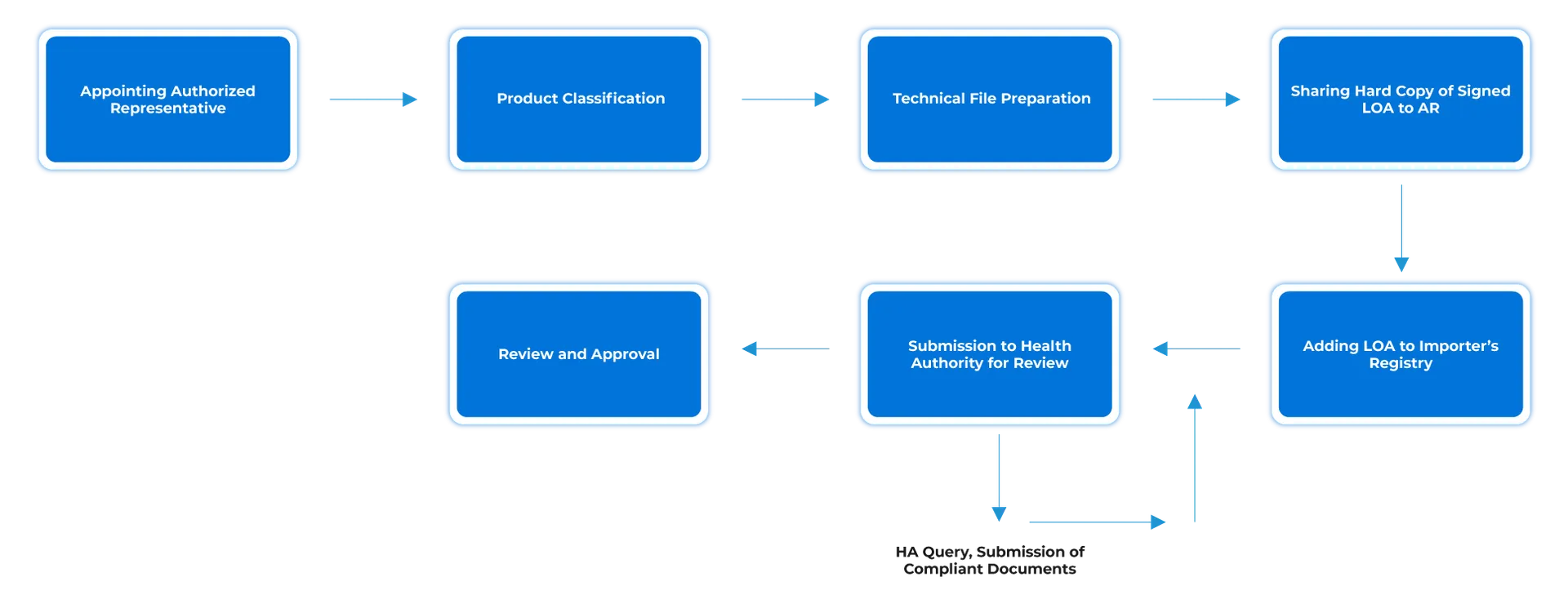
Zarządzanie cyklem życia wyrobu po zatwierdzeniu
Freyr oferuje kompleksowe wsparcie zagranicznym producentom w zarządzaniu całym cyklem życia wyrobów medycznych w Egipcie, w tym działań po zatwierdzeniu:
- Zarządzanie zmianami po zatwierdzeniu, obejmujące modyfikacje istniejących zatwierdzeń wyrobów medycznych, takie jak dodawanie nowych wariantów, akcesoriów i wskazań do stosowania.
- Utrzymywanie ISO 13485:2016.
- Certyfikacja CE.
- Pełnienie roli pośrednika między Jednostką Notyfikowaną (NB) a producentem.
- System nadzoru mający na celu monitorowanie bezpieczeństwa wyrobu medycznego po uzyskaniu pozwolenia na dopuszczenie do obrotu produktu leczniczego.
- Dostarczać okresowe aktualizacje dotyczące bezpieczeństwa i skuteczności wyrobu medycznego, a także wszelkich zmian w statusie regulacyjnym w innych jurysdykcjach.
- Odnowienie zezwolenia na wprowadzenie do obrotu, w zależności od rodzaju wyrobu i przepisów, po określonym czasie.
Skuteczne zarządzanie nadzorem po wprowadzeniu do obrotu (PMS) w Egipcie wymaga umiejętnego poruszania się po ramach regulacyjnych ustanowionych przez EDA. Podmioty wchodzące na rynek, które borykają się z tymi zawiłościami i nie posiadają ugruntowanego partnera regulacyjnego, mogą skorzystać z szerokiego zakresu usług regulacyjnych oferowanych przez Freyr Solutions. Usługi te przyczyniają się do płynnego procesu zatwierdzania wyrobów medycznych w Egipcie, gwarantując ciągłą zgodność z ciągle zmieniającym się krajobrazem regulacyjnym i dynamiką rynku.
Rejestracja Wyrobów Medycznych w Egipcie – Ekspertyza
- Analiza informacji regulacyjnych.
- Należyta staranność regulacyjna.
- Klasyfikacja wyrobów medycznych.
- Rejestracja wyrobów.
- Egipski Posiadacz Rejestracji.
- Wsparcie tłumaczeniowe.
- Pisanie tekstów medycznych.
- Wsparcie w zakresie etykietowania.
- Identyfikacja i kwalifikacja dystrybutora.
- Zarządzanie zmianami po zatwierdzeniu.
- Odnowienie i przeniesienie licencji.
- Odprawa celna.
