
Przegląd rejestracji wyrobów medycznych w Izraelu
Izraelska branża wyrobów medycznych doświadcza stałego wzrostu i innowacji, stając się centrum najnowocześniejszych technologii medycznych. Rejestracja wyrobów medycznych jest kluczowa dla firm wchodzących na ten dynamiczny rynek. Niniejszy przegląd przedstawia kluczowe aspekty izraelskiego procesu rejestracji, oferując wgląd w ramy regulacyjne i wymagania dotyczące wprowadzania innowacyjnych wyrobów medycznych na czoło izraelskiego sektora opieki zdrowotnej.
Organ regulacyjny: Wydział Wyrobów Medycznych Izraelskiego Ministerstwa Zdrowia (AMAR).
Regulacja: Ustawa o Wyrobach Medycznych/Sprzęcie Medycznym z 2012 r.
Ścieżka regulacyjna: Rejestracja produktu
Lokalny Autoryzowany Przedstawiciel w Izraelu: Posiadacz Rejestracji w Izraelu (IRH)
Wymóg QMS: ISO 13485
Ocena danych technicznych: Departament Wyrobów Medycznych w Ministerstwie Zdrowia
Ważność licencji: Pięć (05) lat
Format składania: Papierowy i elektroniczny
Tłumaczenie: Przetłumaczone dokumenty w języku hebrajskim
Klasyfikacja wyrobów
Prawo dotyczące Sprzętu Medycznego i Rozporządzenia dotyczące Rejestracji Sprzętu Medycznego w Izraelu nie określają systemu klasyfikacji ryzyka. Zamiast tego, Izrael dostosowuje swoją klasyfikację wyrobów medycznych do standardów międzynarodowych, w szczególności tych określonych przez kraje Global Harmonization Task Force (GHTF). Alternatywnie, klasyfikacja ryzyka wyrobu w uznanym kraju jest przyjmowana do rejestracji w Izraelu. Ten proces klasyfikacji zazwyczaj uwzględnia zamierzone zastosowanie, poziom ryzyka i inne czynniki, które mogą wpływać na bezpieczeństwo i skuteczność wyrobów medycznych.
Klasy wyrobów medycznych
| Klasa | Ryzyko |
|---|---|
| Klasa I | Niski |
| Klasa II | Nisko-średni |
| Klasa III | Wysoki |
Proponowane zmiany w ścieżkach rejestracji
Proponowane modyfikacje dotyczą wyrobów klasy I i klasy II, natomiast system rejestracji wyrobów klasy III pozostaje bez zmian.
- Urządzenia klasy I mogą być natychmiast zarejestrowane poprzez samodeklarację.
- W przypadku wyrobów klasy II, choć deklaracje i dokumenty techniczne są niezbędne, AMAR może przyspieszyć proces do czternastu (14) dni dla tych uznanych za niskie lub średnie ryzyko. Dotyczy to sytuacji, gdy producent posiada dwa (02) upoważnienia z uznanych krajów i dostarcza dane rynkowe z sześciu (06) miesięcy. Alternatywnie, dla wyrobów klasy II posiadających jedynie zezwolenie US FDA 510(k) i sześć (06) miesięcy danych rynkowych z US, czas przetwarzania AMAR jest przyspieszony do sześćdziesięciu (60) dni.
Lokalny upoważniony przedstawiciel w Izraelu
Firmy produkujące wyroby medyczne, mające siedzibę poza Izraelem, muszą wyznaczyć izraelskiego posiadacza rejestracji (IRH), aby ułatwić rejestrację swoich produktów do sprzedaży w tym kraju. IRH działa jako lokalny przedstawiciel producenta i jest odpowiedzialny za współpracę z Ministerstwem Zdrowia w celu zapewnienia zgodności z lokalnymi przepisami. Ponadto, IRH odpowiada za ustanowienie i utrzymanie obecności biznesowej w Izraelu, a także za uzyskanie i utrzymanie ważnej licencji handlowej.
Rejestracja wyrobów medycznych
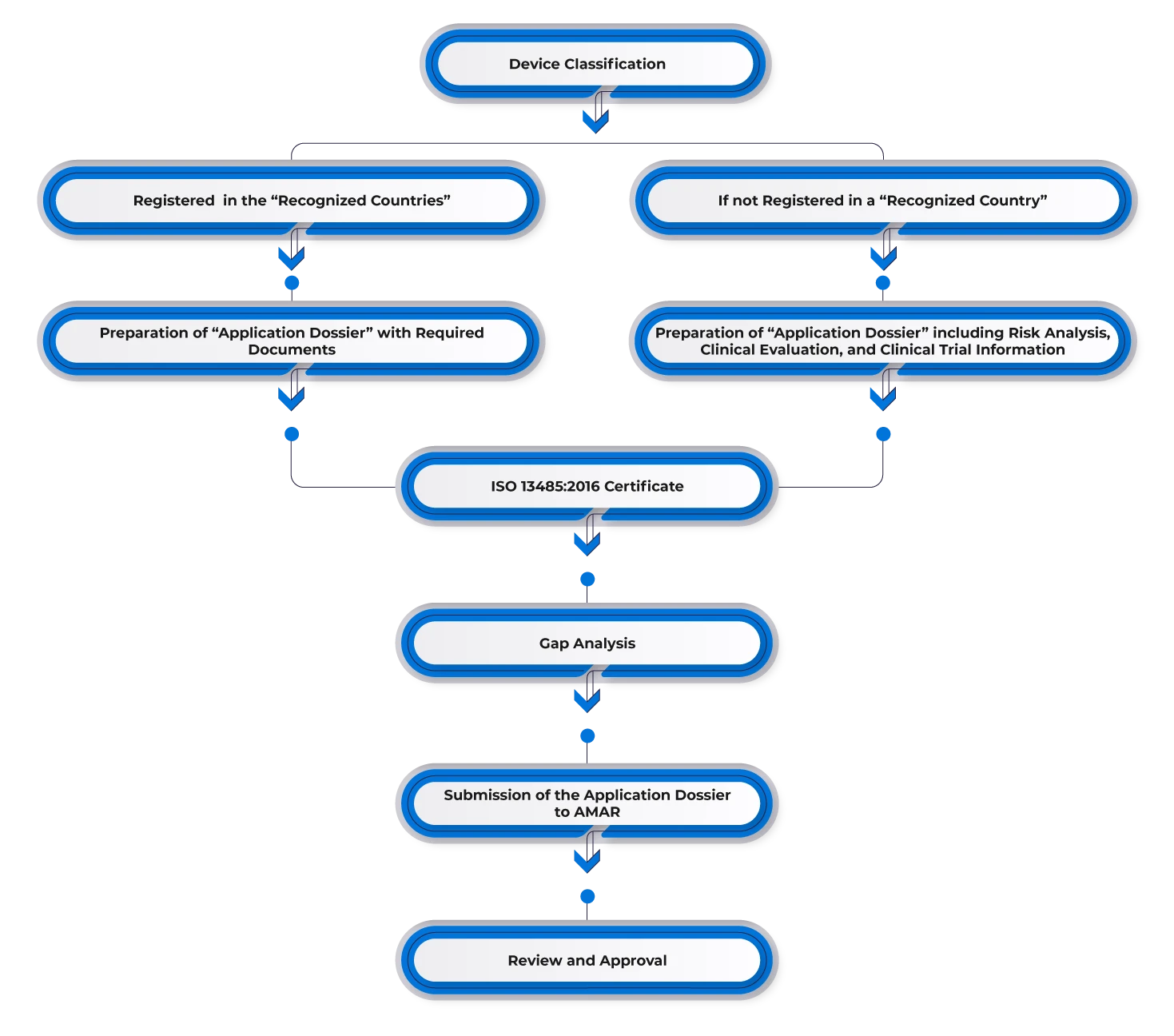
Aby zarejestrować wyrób medyczny w Izraelu, producenci muszą uzyskać wcześniejsze zatwierdzenie na jednym z rynków referencyjnych, takich jak USA, Europa, Australia, Kanada lub inne główne rynki. Producenci posiadający istniejące zatwierdzenia w jednym z krajów referencyjnych mogą wykorzystać to zatwierdzenie na rynku izraelskim i wyznaczyć przedstawiciela w kraju. Następnie muszą przedłożyć wymaganą dokumentację, w tym:
- FDA 510(k)/Pismo zatwierdzające przed wprowadzeniem do obrotu/CE.
- Certyfikat dla Rządu Zagranicznego (CFG)/Certyfikat Wolnej Sprzedaży (CFS).
- ISO 13485 lub inna uznana certyfikacja Dobrych Praktyk Wytwarzania (GMP).
- Walidacja i certyfikacja przez Instytut Norm Izraela (w razie potrzeby).
Przebieg procesu
Zarządzanie cyklem życia wyrobu po zatwierdzeniu
Freyr oferuje kompleksowe wsparcie zagranicznym producentom w zarządzaniu całym cyklem życia wyrobów medycznych w Izraelu, w tym działań po zatwierdzeniu:
- Zarządzanie zmianami po zatwierdzeniu, obejmujące modyfikacje istniejących zatwierdzeń wyrobów medycznych, takie jak dodawanie nowych wariantów, akcesoriów i wskazań do stosowania.
- Utrzymanie certyfikacji ISO 13485:2016 i CE.
- Odnowienie licencji.
- Pełnienie roli pośrednika między Jednostką Notyfikowaną (NB) a producentem.
Poruszanie się po zawiłościach organów autoryzacyjnych i przestrzeganie wielu zestawów przepisów dotyczących zatwierdzania wyrobów może być wyzwaniem. Uzyskanie zatwierdzeń od różnych krajów GHTF i przestrzeganie przepisów stanowych wymaga dogłębnej wiedzy regulacyjnej. Dla podmiotów wchodzących na rynek, które borykają się z tymi złożonościami bez ustalonego partnera regulacyjnego, Freyr oferuje kompleksowe usługi regulacyjne End-to-End, usprawniając proces zatwierdzania wyrobów medycznych w Izraelu.
Rejestracja Wyrobów Medycznych w Egipcie – Ekspertyza
- Klasyfikacja wyrobów medycznych w Izraelu.
- Posiadacz rejestracji w Izraelu (IRH).
- Rejestracja wyrobów w Izraelu.
- ISO 14971:2019 Konsultacje w zakresie zarządzania ryzykiem.
- Zgodność z ISO 13485:2016.
- Przegląd, opracowanie i złożenie dokumentacji projektowej.
- Rejestracja wyrobów medycznych za pośrednictwem internetowego systemu rejestracji.
- Raport ze strategii regulacyjnej wyrobów medycznych.
- Wsparcie w badaniach – biokompatybilność, bezpieczeństwo elektryczne, właściwości mechaniczne i użytkowe.
- Wsparcie w zakresie zgodności oznakowania.
- Wsparcie GMP.
- Wsparcie w zakresie nadzoru po wprowadzeniu do obrotu (PMS).
