
Opis zakresu prac (SOW) dotyczący wniosku 510(k) w odniesieniu do aktywnych i nieaktywnych wyrobów medycznych – przegląd
W firmie Freyr nasz zespół ekspertów sumiennie gromadzi i selekcjonuje najnowsze informacje niezbędne do przygotowania wniosków 510(k), obejmujące zarówno urządzenia aktywne, jak i nieaktywne wyroby medyczne. Dzięki temu zyskujesz wiedzę niezbędną do pewnego poruszania się po ramach regulacyjnych. Od wyjaśnienia różnic między wyrobami aktywnymi a nieaktywnymi po dogłębną analizę zawiłości procesu składania wniosków 510(k) – stworzyliśmy obszerne repozytorium zasobów, które posłuży Państwu jako podstawowe źródło informacji. Zapraszamy do zapoznania się z naszym kompleksowym przewodnikiem, który pomoże Państwu opanować proces składania wniosków 510(k) dotyczących aktywnych wyrobów medycznych oraz nieaktywnych wyrobów medycznych wyroby medyczne .
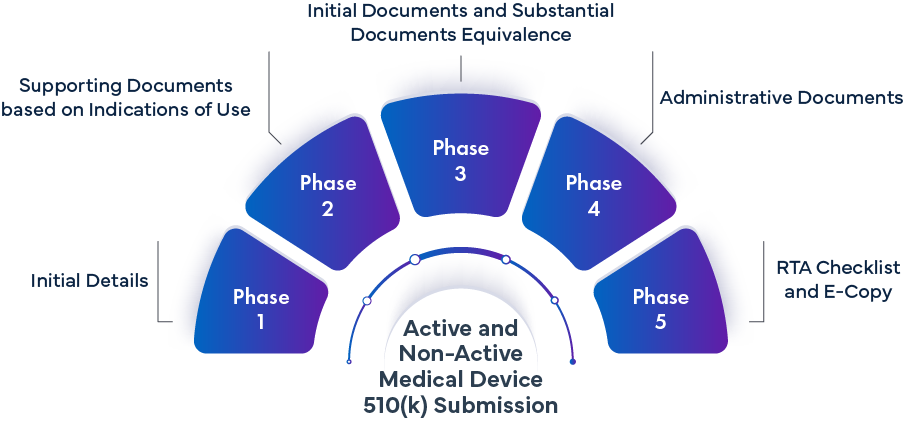
Faza -1: Wstępne szczegóły | ||
|---|---|---|
Wymogi | Zakres dla wnioskodawcy 510(k) | Zakres działalności Freyr |
| Przeznaczenie |
|
|
| Oświadczenie o przeznaczeniu (Formularz 3881) |
|
|
| Opis wyrobu |
|
|
| Standardy i wytyczne |
|
|
| Urządzenie referencyjne |
|
|
| Podsumowanie dotyczące procedury 510(k) |
|
|
Faza 2: Dokumentacja pomocnicza na podstawie wskazań do stosowania | |||
|---|---|---|---|
Wymogi dokumentacyjne | Zakres dla wnioskodawcy 510(k) | Zakres działalności Freyr | |
| 2.1 | Rysunek wyrobu | Prześlij plik rysunkowy urządzenia, aby zapewnić dokładne przedstawienie jego projektu. | Zainicjuj formalny wniosek o rysunek aktywnego urządzenia. Dokładnie przejrzyj i skrupulatnie udokumentuj niezbędne informacje do zgłoszenia 510(k). |
| 2.2 | Projektowanie i rozwój urządzenia | Prześlij plik projektu i rozwoju aktywnego urządzenia, zawierający wszystkie istotne informacje i dokumentację. | Złóż wniosek o projekt i rozwój aktywnego urządzenia. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.3 | Karta charakterystyki substancji niebezpiecznej | Dostarczyć Karty Charakterystyki Substancji Niebezpiecznych (MSDS) dla kluczowych komponentów aktywnego urządzenia, zapewniając kompleksowe informacje dotyczące ich bezpieczeństwa i składu. | Wyślij zapytanie o kartę charakterystyki kluczowych komponentów aktywnego urządzenia. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.4 | Schemat procesu produkcyjnego | Dostarcz schemat przepływu produkcji, szczegółowo opisujący proces produkcji aktywnego urządzenia, zapewniający wizualne przedstawienie etapów produkcji i ich kolejności. | Złóż wniosek o Kartę Charakterystyki Substancji Niebezpiecznej (MSDS) podstawowych komponentów dla aktywnego urządzenia. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.5 | Opis wyrobu | Dostarczyć wyczerpujące szczegóły, w tym: o Przegląd urządzenia o Funkcje i tryby działania o Schematy blokowe o Zdjęcia, kable i odpowiednie akcesoria o Interoperacyjność urządzenia. o Opis zasilania | Złóż wniosek o szczegółowe informacje dotyczące urządzenia. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.6 | Proponowane etykietowanie | Dostarczyć instrukcję użytkowania (IFU), instrukcję obsługi oraz wszelkie powiązane materiały promocyjne dla aktywnego urządzenia. | Złóż wniosek o Instrukcję Używania (IFU), Instrukcję Obsługi oraz wszelkie materiały promocyjne, jeśli są dostępne. Przejrzyj IFU, instrukcję obsługi i materiały promocyjne dostarczone przez wnioskodawcę. Udokumentuj IFU, instrukcję obsługi i materiały promocyjne w celu złożenia wniosku 510(k). |
| 2.7 | Opakowania i Transport | Dostarczyć plany badań i raporty dotyczące walidacji opakowania i transportu. | Złóż wniosek o plan badania i raporty dotyczące walidacji opakowania i transportu. Przejrzyj plany badań i raporty dotyczące walidacji opakowania i transportu oraz przedstaw wszelkie niezbędne poprawki lub uwagi. |
| 2.8 | Sterylizacja (jeśli wymagana jest sterylność) | Dostarczyć plany badań i raporty dla walidacji sterylizacji. | Złożyć wniosek o plan badania walidacji sterylizacji i raporty. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.9 | Testowanie wydajności _ Stanowisko testowe | Zainicjuj formalny wniosek o plany i raporty z badań laboratoryjnych dotyczących testów wydajności, określające konkretne wymagania i cele do realizacji. | Złożyć wniosek o plany i raporty z badań laboratoryjnych aktywnego wyrobu w celu testowania wydajności. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
Dokumentacja uzupełniająca dotycząca kompatybilności elektromagnetycznej i bezpieczeństwa elektrycznego | |||
| 2.10 | Charakterystyka urządzeń związana z EMC i środowiska zamierzonego użytkowania | Dostarczyć szczegółowe informacje dotyczące charakterystyki wyrobu związanej z EMC oraz środowisk zamierzonego zastosowania, w tym: o Przegląd urządzenia. o Funkcje i tryby działania. o Schematy blokowe. o Zdjęcia, kable i odpowiednie akcesoria. o Interoperacyjność urządzenia. o Opis zasilania, w tym możliwości używania wewnętrznie zasilanego wyrobu medycznego podczas ładowania. o Środowiska, w których wyrób medyczny ma być używany. o Opis wszelkich technologii bezprzewodowych (jeśli dotyczy) dla dodatkowych uwag dotyczących wyrobów medycznych z obsługą bezprzewodową. o Opis wszelkich wewnętrznych emiterów RF w wyrobie medycznym, które potencjalnie mogłyby powodować zakłócenia elektromagnetyczne. o Omówienie powszechnych emiterów elektromagnetycznych (EM), jak również unikalnych emiterów medycznych.
| Złożyć wniosek o informacje dotyczące charakterystyki wyrobu związanej z EMC oraz środowisk zamierzonego zastosowania. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.11 | Ocena ryzyka | Dostarczyć Plan Zarządzania Ryzykiem, który zawiera ocenę ryzyka prezentującą skuteczne łagodzenie ryzyka, wraz z kompleksowym raportem zarządzania ryzykiem obejmującym wszystkie elementy ryzyka. Dostarczyć poprawiony dokument z sugerowanymi poprawkami i ulepszeniami | Złożyć wniosek o Plik Zarządzania Ryzykiem i zażądać dokumentacji Planu i Raportu Zarządzania Ryzykiem, w tym identyfikacji zagrożeń ryzyka, oceny ryzyka oraz wykazania odpowiedniego łagodzenia ryzyka. Raport Zarządzania Ryzykiem powinien obejmować wszystkie elementy ryzyka, najlepiej z osobnymi sekcjami dla jasności. Dostarczyć szablon Planu Zarządzania Ryzykiem oraz Raportu Zarządzania Ryzykiem, obejmujący wszystkie ryzyka związane z wyrobem, na żądanie wnioskodawcy. Przejrzeć dane w Pliku Zarządzania Ryzykiem, w tym Plan i Raport udostępnione przez wnioskodawcę, oraz przedstawić sugestie dotyczące niezbędnych poprawek, aby zapewnić kompleksową dokumentację do złożenia wniosku 510(k). Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.12 | Norma konsensusowa | Dostarczenie potwierdzenia odpowiednich norm konsensusowych oraz wyjaśnienia wszelkich odstępstw od norm uznanych przez FDA. | Złożyć wniosek o obowiązujące normy konsensusowe związane z EMC i bezpieczeństwem elektrycznym dla aktywnego wyrobu. Udokumentuj potwierdzone normy konsensusu dla aktywnego wyrobu w celu złożenia zgłoszenia 510(k). |
| 2.13 | Niezbędne kryteria zaliczenia/niezaliczenia w zakresie wydajności i odporności | Prześlij plan badania i raporty z testów Podstawowej Wydajności i Odporności przeprowadzonych na aktywnym urządzeniu, zgodnie ze standardami uznanymi przez FDA. | Złóż zapotrzebowanie na plan badania i raporty z testów Podstawowej Wydajności i Odporności przeprowadzonych na aktywnym urządzeniu, zgodnie ze standardami uznanymi przez FDA. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.14 | Konfiguracja i testowane funkcje wyrobu medycznego | Dostarczyć konfigurację wyrobu medycznego i przetestowane funkcje dla aktywnego wyrobu, obejmujące następujące szczegóły: o Przedstawić szczegółowy opis badanego wyrobu medycznego, zawierający szczegółowe informacje na temat jego konfiguracji, funkcji, trybów pracy oraz konkretnych testowanych ustawień. o Opis badanego wyrobu powinien zawierać nazwę wyrobu medycznego, numer modelu oraz wskazywać, czy wyrób jest ostatecznym, gotowym do produkcji wyrobem medycznym, który jest obecnie w trakcie oceny. | Złożyć wniosek o konfigurację i funkcje testowe aktywnego wyrobu medycznego. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
| 2.15 | Wyniki testów EMC | Dostarczyć plan i raport z badań EMC (kompatybilności elektromagnetycznej) zgodnie z uznanym przez FDA standardem konsensusu, zalecanym dla aktywnego urządzenia. | Zainicjuj formalny wniosek o plan badania i raport z testów EMC, zgodny z uznanym przez FDA standardem konsensusu zalecanym dla aktywnych wyrobów. Dokładnie przejrzyj i skrupulatnie udokumentuj wszystkie niezbędne informacje w ramach przygotowań do zgłoszenia 510(k). |
Faza 3 – Dokumenty wstępne i dokumenty dotyczące równoważności zasadniczej | |||
|---|---|---|---|
Wymogi dokumentacyjne | Zakres dla wnioskodawcy 510(k) | Zakres działalności Freyr | |
| 3.1 | Arkusz informacyjny zgłoszenia do przeglądu przed wprowadzeniem na rynek CDRH (Formularz FDA 3514) | - | Wypełnij formularz FDA 3514, korzystając z danych dostarczonych przez wnioskodawcę |
| 3.2 | Podsumowanie i certyfikacja klasy III | - | Ten krok nie jest konieczny, jeśli badania kliniczne nie są wymagane. |
| 3.3 | Certyfikat finansowy lub oświadczenie o ujawnieniu informacji | - | Ten krok nie jest konieczny, jeśli badania kliniczne nie są wymagane. |
| 3.4 | Streszczenie wykonawcze | - | Opracuj szablon i starannie przygotuj dokument. Dostarczyć uzasadnienia dla wszelkich rozbieżności zaobserwowanych między proponowanym wyrobem a wyrobem referencyjnym. Wybiera się studium porównawcze między proponowanym wyrobem a wyrobem referencyjnym, tworzy szablon i przygotowuje odpowiedni dokument. |
| 3.5 | Omówienie istotnej równoważności | - | Opracuj szablon i starannie przygotuj dokument. Wybiera się studium porównawcze między proponowanym wyrobem a wyrobem referencyjnym, tworzy szablon i przygotowuje odpowiedni dokument. |
Faza 4 – Dokumenty administracyjne | |||
|---|---|---|---|
Wymogi dokumentacyjne | Zakres dla wnioskodawcy 510(k) | Zakres działalności Freyr | |
| 4.1 | List przewodni 510(k) | Podpisz dokument wydrukowany na papierze firmowym i zorganizuj wysyłkę papierowej kopii kurierem do biura w US. Dostarczyć cyfrową kopię podpisanego listu przewodniego 510(k) do włączenia do dokumentacji 510(k). | Przygotowanie kompleksowego szablonu zawierającego wszystkie niezbędne szczegóły dla listu przewodniego i przekazanie go wnioskodawcy. Poinstruowanie wnioskodawcy, aby użył swojego oficjalnego papieru firmowego oraz zapewnienie, że list przewodni jest podpisany przez upoważnioną osobę. |
| 4.2 | Oświadczenie o prawdziwości i dokładności | Zapewnij, że dokument jest podpisany przez wyznaczoną osobę kontaktową w firmie i dostarczony zgodnie z wymogami. | Opracuj kompleksowy szablon zawierający całą niezbędną treść do uwzględnienia w dokumencie zgłoszeniowym. |
| 4.3 | Deklaracje zgodności i Raport podsumowujący | Zapewnij, że dokument jest podpisany przez wyznaczoną osobę kontaktową w firmie i dostarczony zgodnie z wymogami. | Opracuj kompleksowy szablon do systematycznego sporządzania listy i przygotowywania wymaganych dokumentów. |
| 4.4 | MDFUSC (Formularz FDA 3601) | Dokonaj wymaganej płatności do FDA przed formalnym zgłoszeniem pliku 510(k). | Wygeneruj stronę tytułową opłaty użytkownika oraz unikalny Numer Identyfikacji Osobistej (PIN) specjalnie dla zgłoszenia wyrobu medycznego. |
Faza 5 – Lista kontrolna RTA i kopia elektroniczna | |||
|---|---|---|---|
Wymogi dokumentacyjne | Zakres dla wnioskodawcy 510(k) | Zakres działalności Freyr | |
| 5.1 | Lista kontrolna RTA | Zatwierdzenie weryfikacji listy kontrolnej RTA (Gotowe do Akceptacji), wskazujące, że wszystkie wymagania zostały pomyślnie spełnione. | Opracuj dostosowany szablon listy kontrolnej RTA, dostosowany do konkretnego typu zgłoszenia. Uzupełnij listę kontrolną, starannie wypełniając wszystkie wymagane pola i upewniając się, że wymienione dokumenty zostały należycie złożone do FDA i udostępnione wnioskodawcy. |
| 5.2 | E-kopia | Zatwierdzenie dokumentacji zawartej w folderze ostatecznego złożenia, potwierdzające jej zgodność ze wszystkimi niezbędnymi wymaganiami i normami. | Uporządkuj sekcje folderu zgłoszeniowego zgodnie z wytycznymi FDA i niezwłocznie udostępnij je wnioskodawcy. Przekształć folder zgłoszeniowy w kopię elektroniczną, aby zapewnić wygodny dostęp i możliwość przeglądu. Prześlij elektroniczną kopię zgłoszenia wyznaczonemu agentowi w US. |
Rejestracja wyrobów medycznych
- Kompleksowa strategia regulacyjna US FDA.
- Identyfikacja urządzenia referencyjnego
- Ustanowienie istotnej równoważności z urządzeniem referencyjnym
- Analiza luk dla zgodności z US FDA
- Opracowanie 21 sekcji pliku technicznego 510(k)
- Publikowanie i tworzenie e-kopii
- Walidacja i złożenie eCopy.
- Usługi pośrednictwa w celu zatwierdzenia wyrobu
- Rozpatrzenie odpowiedzi RTA i niedociągnięć
- Usługi konsultacyjne w zakresie usuwania braków
- Wykaz urządzeń i utrzymanie bazy danych FURLS

- Obsługiwaliśmy wiele rejestracji 510(k) dla zróżnicowanych kategorii wyrobów medycznych.
- Zespół ekspertów do kompilacji 510(k) zgodnie z wymaganiami US FDA dotyczącymi zgłoszenia przed wprowadzeniem do obrotu (510(k))
- Dodatkowe wsparcie w obsłudze zapytań dotyczących 510(k)
- Doradztwo w zakresie odpowiedniego typu zgłoszenia 510(k) zgodnie z wymogami zgłoszeniowymi US FDA 510(k) dla wyrobu.
- Terminowe złożenie rezultatów
- Na bieżąco z nowymi poprawkami US FDA
