Przegląd przedrynkowego zatwierdzania urządzeń medycznych USFDA
Proces zatwierdzania USFDA (PMA) USFDA jest jedną z procedur rejestracji wyrobów medycznych przewidzianych przez US FDA, przeznaczoną przede wszystkim dla wyroby medyczne FDA III. Proces zatwierdzania FDA dla wyrobów klasy III obejmuje skrupulatną ocenę naukową i regulacyjną w celu oceny bezpieczeństwa i skuteczności wyrobu medycznego, zapewniając spełnienie najwyższych standardów przed dopuszczeniem do obrotu.
Zarezerwuj spotkanie z naszymi ekspertami ds. zatwierdzeń przed wprowadzeniem do obrotu
Who powinien złożyć wniosek USFDA o zatwierdzenie wyrobu medycznego przed wprowadzeniem do obrotu (PMA)?
Producenci urządzeń muszą złożyć wniosek PMA, jeśli urządzenie:
- To powieść.
- Należy do klasy wysokiego ryzyka.
- Nie można znaleźć w bazie danych klasyfikacji produktów.
- Nie jest zasadniczo równoważny (NSE) z urządzeniami klasy I, II lub III.
Uzyskaj porady ekspertów dotyczące wniosku o zatwierdzenie przed wprowadzeniem do obrotu
Jaka jest różnica między wnioskami 510(k), PMA i De-Novo?
Zatwierdzenie przed wprowadzeniem do obrotu
- Wyrób należący do klasy III, który podtrzymuje życie ludzkie lub stwarza potencjalne, nieuzasadnione ryzyko choroby lub urazu.
- Proces zatwierdzania PMA FDA wymaga przeprowadzenia badań klinicznych.
- Wymaga kontroli na miejscu przed wydaniem zatwierdzenia PMA.
- 180 dni kalendarzowych
Klasyfikacja De-Novo
- Nowe wyroby klasy I i II, które nie mają ważnego wyrobu źródłowego.
- Wymaga danych z badań klinicznych.
- Brak audytu na miejscu przed zatwierdzeniem De-Novo.
- 150 dni kalendarzowych.
Rejestracja 510(k)
- Urządzenia klasy III FDA , które są zasadniczo równoważne z urządzeniem źródłowym.
- Nie wymaga testów na ludziach.
- Brak audytu na miejscu przed zatwierdzeniem 510(k).
- 90 dni kalendarzowych.
Jakie są różne metody składania wniosków o zatwierdzenie przed wprowadzeniem na rynek FDA ?
Producenci mogą wybrać jedną z następujących czterech (04) metod aplikacji PMA, która będzie najbardziej odpowiednia dla ich urządzenia:
- Tradycyjne PMA
- Modułowy PMA
- Protokół rozwoju produktu
- Zwolnienie z obowiązku stosowania urządzeń humanitarnych
Jakie są wymagania dotyczące danych dla zatwierdzania wyrobów medycznych przed wprowadzeniem do obrotu?
Zgodnie z 21 CFR część 814 wnioskodawcy muszą przedłożyć US agencjiFDA należycie wypełniony formularz wniosku CDRH, wymagane zobowiązania oraz dobrze sporządzoną dokumentację techniczną PMA. Dokumentacja techniczna powinna zawierać dane niekliniczne i kliniczne.
Dane niekliniczne - obejmują dane dotyczące mikrobiologii, toksykologii, immunologii, biokompatybilności, stresu, zużycia, okresu trwałości i innych testów laboratoryjnych lub testów na zwierzętach.
Dane kliniczne - obejmują dane dotyczące protokołów badań, dane dotyczące bezpieczeństwa i skuteczności, działania niepożądane i powikłania, awarie i wymiany urządzeń, informacje o pacjentach, skargi pacjentów, zestawienia danych od wszystkich poszczególnych uczestników, wyniki analiz statystycznych i wszelkie inne informacje z badań klinicznych.
Jak wygląda proces aplikacji PMA?
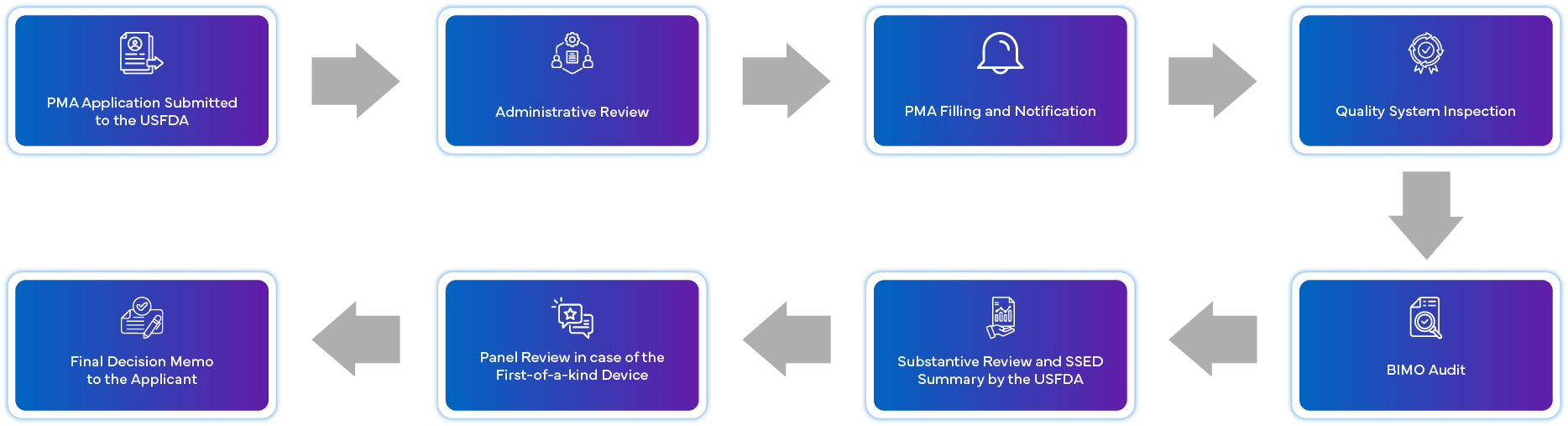
Jakie są wymagania dotyczące zgodności po zatwierdzeniu dla PMA?
Urządzenia zatwierdzone w ramach ścieżki PMA muszą spełniać wymogi po wprowadzeniu do obrotu określone przez USFDA. Urządzenie musi spełniać następujące wymogi:
- Wymogi po zatwierdzeniu nałożone w nakazie zatwierdzenia FDA PMA.
- Zarządzanie zmianami po zatwierdzeniupoprzez terminowe składanie odpowiednich suplementów PMA
- Składanie raportów po zatwierdzeniu (rocznych)
- Przepisy 21 CFR 803 dotyczące raportowania wyrobów medycznych (MDR)
- Badania nadzoru po wprowadzeniu do obrotuwymagane przez USFDA w nakazach zatwierdzenia PMA.
Jakie są opłaty USFDA za rozpatrzenie wniosku PMA?
Opłaty dla użytkowników MDUFA za oryginalne PMA i suplementy są następujące-
| Typ aplikacji | Opłaty za rok podatkowy 2023 (od sup października | |
|---|---|---|
| Opłata standardowa | Opłata dla małych firm | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Dodatek Panel-Track | $353,238 | $88,309 |
| 180-dniowy dodatek | $66,232 | $16,558 |
| Opłata roczna za okresowe raportowanie dotyczące urządzenia klasy III (PMA, PDP i PMR) | $15,454 | $3,864 |
| 30-dniowe powiadomienie | $7,065 | $3,532 |
| Dodatek w czasie rzeczywistym | $30,908 | $7,727 |
Dzięki doświadczeniu w obsłudze zgłoszeń PMA, Freyr może pomóc w identyfikacji i kompilacji informacji oraz pomóc w przygotowaniu i przeglądzie wniosku.
Wiedza i zalety USFDA w zakresie zatwierdzania wyrobów medycznych przed wprowadzeniem na rynek
- Należyta staranność regulacyjna
- Zgodność z inspekcją systemu jakości
- Zgodność z wymogami audytu BIMO
- Kompilacja plików technicznych PMA
- Publikowanie i tworzenie e-kopii
- Zatwierdzanie i składanie eKopii
- Odpowiedzi i niedociągnięcia RTA
- Usługi łączności do czasu zatwierdzenia przez FDA przed wprowadzeniem na rynek
- Konsultacje w sprawie niedociągnięć
- Lista urządzeń i rejestracja zakładów
- Zarządzanie suplementami PMA i 30-dniowymi powiadomieniami
- Składanie rocznych raportów okresowych
- Próbne audyty i 21 CFR 820 Szkolenie

- Doświadczenie w obsłudze wielu zgłoszeń FDA PMA dla zróżnicowanych kategorii urządzeń.
- Zespół ekspertów ds. wniosków o zatwierdzenie przed wprowadzeniem do obrotu FDA zgodnie z wymogami regulacyjnymi
- Dodatkowe wsparcie w zakresie obsługi zapytań związanych z PMA.
- Terminowe przekazywanie wyników
- Zgodność zFDA zmianami wprowadzonymi przez US agencjęFDA
