4 min read
Zgłaszanie wyrobów medycznych (MDR) to narzędzie nadzoru po wprowadzeniu do obrotu, wykorzystywane przez Agencję Żywności i LekówFDA) do monitorowania wydajności urządzeń, wykrywania potencjalnych kwestii bezpieczeństwa związanych z urządzeniami i przyczyniania się do oceny korzyści i ryzyka związanego z urządzeniami. Celem MDR jest wykrywanie i reagowanie na zdarzenia niepożądane związane z urządzeniami w odpowiednim czasie. Umożliwia on lekarzom, placówkom opieki zdrowotnej, producentom i konsumentom dobrowolne zgłaszanie zdarzeń w celu zrozumienia bezpieczeństwa i skuteczności urządzenia po jego wprowadzeniu do obrotu.
MDR ma zastosowanie do wszystkich klas wyroby medyczne, które są produkowane w Stanach Zjednoczonych Ameryki (USA) lub importowane do USA. Producenci wyrobów medycznych, którzy chcą wprowadzać swoje wyroby do obrotu w USA, muszą przestrzegać przepisów MDR, w przeciwnym razie mogą zostać na nich nałożone kary finansowe. Rozporządzenie ma zastosowanie w USA, w tym w przypadku zdarzeń zagranicznych, tzn. ma zastosowanie do wyroby medyczne legalnie wprowadzonych do obrotu wyroby medyczne Stanach Zjednoczonych, zarówno tych produkowanych w USA, jak i w innych krajach. Ponadto istnieją różne przypadki stosowania MDR, takie jak:
- jeśli urządzenie jest produkowane w USA, dystrybuowane lokalnie i na inne rynki
- gdy urządzenie jest produkowane w USA, ale dystrybuowane na innych rynkach
- gdy urządzenie jest produkowane w obcym kraju, dostarczane do USA i na inne rynki
- gdy urządzenie jest produkowane w obcym kraju i dystrybuowane lokalnie oraz
- gdy urządzenie jest przedmiotem dochodzenia w USA
MDR i przepływ procesu raportowania
Rozporządzenie MDR zawiera wiele obowiązkowych wymogów dla producentów, importerów i placówek użytkujących urządzenia w zakresie zgłaszania do FDA określonych zdarzeń niepożądanych związanych z urządzeniami i problemów z produktami. Poniższy schemat opisuje krok po kroku proces raportowania.
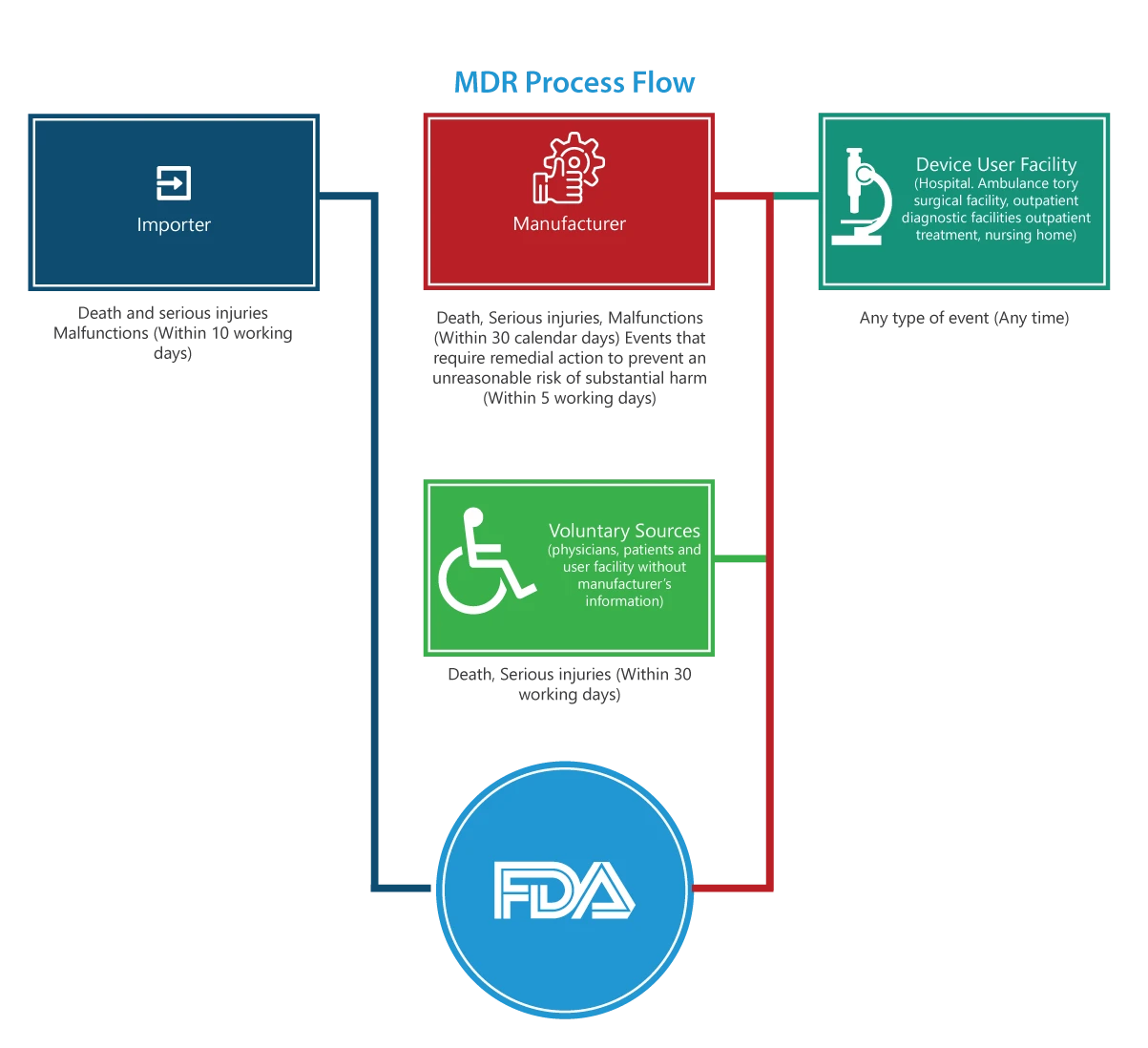
Do kogo ma zastosowanie?
Importerzy
Zgłoszenia zgonów, poważnych obrażeń i nieprawidłowego działania muszą być przekazywane do FDA i producenta w ciągu 30 dni roboczych. Jeśli nieprawidłowe działanie może spowodować obrażenia lub śmierć w innym miejscu, importerzy muszą zgłosić nieprawidłowe działanie producentowi.
Producenci
Raporty dotyczące zdarzeń (zgonów, poważnych obrażeń i nieprawidłowego działania) wskazanych przez FDA lub zdarzeń, które wymagają podjęcia działań naprawczych w celu zapobieżenia nieuzasadnionemu ryzyku znacznego uszczerbku na zdrowiu publicznym, muszą zostać przesłane do FDA w ciągu 5 dni roboczych poprzez wypełnienie formularza 3500A.
Placówka użytkownika urządzenia (szpital, ambulatoryjny ośrodek chirurgiczny, dom opieki, ambulatoryjny ośrodek diagnostyczny lub ambulatoryjny ośrodek leczenia)
Raporty muszą być przekazywane producentowi urządzenia nie później niż 10 dni roboczych od dnia, w którym placówka dowiedziała się, że urządzenie spowodowało lub mogło spowodować poważny uraz u pacjenta placówki. Jeśli producent nie jest znany, ośrodek musi przesłać raport do FDA.
Grupy wolontariuszy
Pacjenci, pracownicy służby zdrowia i konsumenci, którzy napotkają problem związany z urządzeniem medycznym, mogą zgłosić się do FDA za pośrednictwem MedWatch
eMDR
W 2015 r. FDA elektronicznego systemu zgłaszania niepożądanych zdarzeń związanych z wyrobami medycznymi (eMDR) w celu identyfikacji krytycznych problemów dotyczących jakości i integralności danych związanych ze zgłaszaniem poważnych urazów związanych ze wszystkimi klasami wyroby medyczne. Preferowaną formą zgłaszania jest eMDR.
Producenci mogą przesyłać swoje eMDR za pośrednictwem elektronicznej bramki zgłoszeńESG). Od momentu przesłania zgłoszenia bramka elektroniczna potrzebuje do 48 godzin na wysłanie potwierdzenia. Jeśli podczas przesyłania raportu wystąpi jakikolwiek błąd, pojawi się komunikat o konieczności wprowadzenia poprawek.
eMDR - jakie korzyści przynosi?
eMDR oferuje wiele korzyści w porównaniu z ręcznym mechanizmem raportowania (tj. MDR). Poniżej wymieniono kilka znaczących korzyści, na które mogą liczyć producenci / agencje / pacjenci:
- Narzędzie do składania wniosków eMDR usprawnia współpracę między organizacją, agencją zdrowiaFDA) i pacjentami.
- eMDR oszczędza koszty. Automatyzacja zmniejsza potrzebę administracyjnych kosztów ogólnych i tradycyjnej komunikacji; pomaga przyspieszyć proces i sprzyja skutecznemu zgłaszaniu zdarzeń, co skutkuje natychmiastową interakcją z FDA.
- Ręczne procesy wymagają znacznej ilości dokumentów, mogą być długotrwałe i trudne do śledzenia i przetwarzania. Przesyłanie eMDR jest zautomatyzowane i scentralizowane. Zapisy można łatwo pobrać, oszczędzając dużo czasu podczas ich przeglądania.
- eMDR umożliwia stronom szybkie oznaczanie błędów w zgłoszeniach, w przeciwieństwie do ręcznej i czasochłonnej korespondencji z FDA
- eMDR działa jako pojedynczy punkt wejścia do przetwarzania wszystkich zgłoszeń elektronicznych w wysoce zabezpieczonym środowisku i jest korzystny, ponieważ skargi w organizacji mogą być bezpośrednio powiązane z formularzem MedWatch i zintegrowane z bramą FDA.
eMDR i przepływ procesu raportowania
Rozporządzenie w sprawie eMDR zawiera obowiązkowe wymogi dla producentów, importerów i placówek użytkujących urządzenia w zakresie zgłaszania do FDA określonych zdarzeń niepożądanych związanych z urządzeniami i problemów związanych z produktami. Poniższy schemat opisuje krok po kroku proces raportowania.
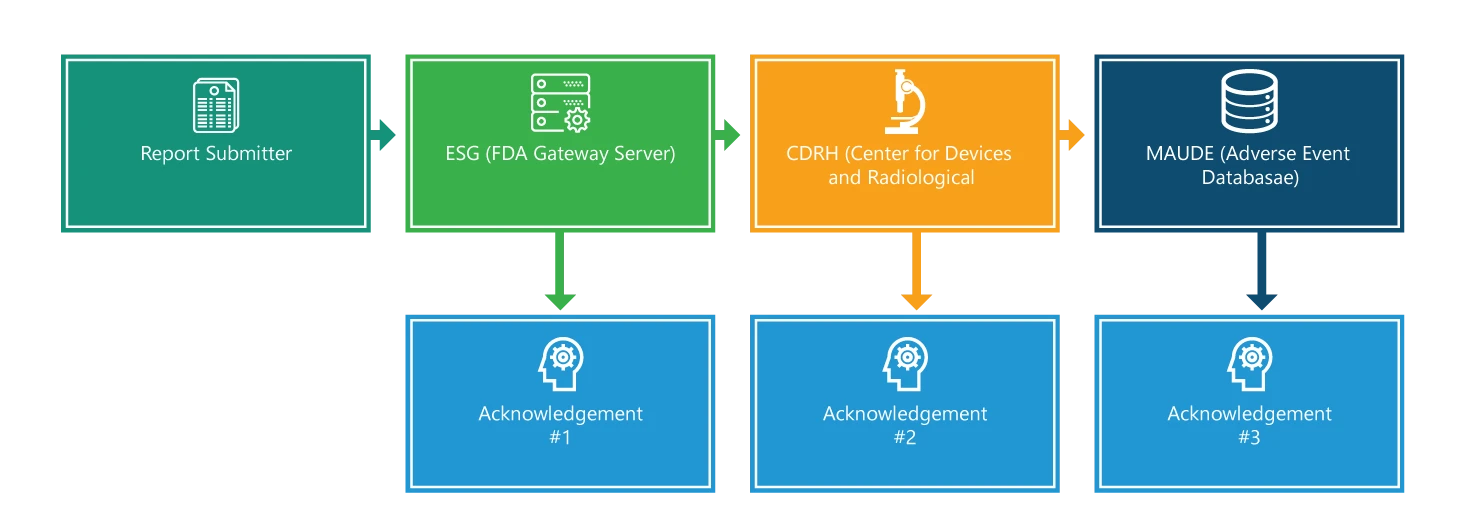
Proces raportowania składa się z czterech kroków. Z wyjątkiem pierwszego kroku, każdy krok jest potwierdzony. Ponadto każdy krok zawiera dodatkowe informacje, które pomogą ułatwić proces.
Krok 1: Zgłaszający raport
Przesyłanie eMDR. Na początku, aby dokonać zgłoszenia, należy posiadać podpis elektroniczny i upewnić się, że nazwy plików zawierają tylko jedną kropkę, która służy do wskazania rozszerzenia typu pliku (na przykład 555xml lub 555.pdf). Czas dostarczenia i przetworzenia aplikacji zależy jednak od całkowitego rozmiaru zgłoszenia; dostarczenie i przetworzenie większych zgłoszeń zajmuje więcej czasu.
Krok 2: Elektroniczna bramka zgłoszeńESG)
Gdy zgłoszenie dotrze do ESG, powinieneś szybko otrzymać potwierdzenie nr 1, chyba że ESG nie działa z powodu konserwacji. Musisz sprawdzić status swojego MDR na stronie internetowej ESG .
Krok 3: CRDH
eMDR jest automatycznie kierowany z ESG do Center for Devices and Radiological Health (CDRH). Po przekierowaniu, tak jak w kroku 2, powinieneś otrzymać potwierdzenie, tj. #2.
Krok 4: Doświadczenie producenta i użytkownika urządzenia (MAUDE)
Gdy CDRH zatwierdzi i zaktualizuje zgłoszenie w bazie danych zdarzeń niepożądanych (MAUDE), oczekuje się, że zgłaszający otrzyma potwierdzenie #3. Należy zauważyć, że wszelkie błędy występujące podczas walidacji i ładowania są rejestrowane.
Raportowanie wyrobów medycznych (MDR) to kluczowy proces, który pomaga ratować życie i chronić pacjentów przed niepotrzebnym ryzykiem. Zapewnia, że wszystkie strony zaangażowane w opiekę nad pacjentem są odpowiedzialne i czujne w korzystaniu z urządzeń.
eMDR ułatwia raportowanie, ale dokumentacja i działania następcze mogą pochłaniać zasoby. Zrób to dobrze za pierwszym razem; porozmawiaj z us na sales@freyrsolutions.com.