2 min read
Głównym celem US FDAjest ciągła kontrola i wypełnianie luk między procesami regulacyjnymi w celu zapewnienia nieprzerwanej importu i sprzedaży nowych i wysokiej jakości wyroby medyczne US .FDA 1998 r. US FDA opublikowała program zatytułowany „Nowy paradygmat 510(k): alternatywne podejścia do wykazywania istotnej równoważności w zgłoszeniach przed wprowadzeniem do obrotu”. Ma on na celu ustanowienie skutecznej ścieżki składania wniosków FDA (k), która zawiera pewne zmiany w już zatwierdzonym wniosku 510(k). To nowe zgłoszenie 510(k) oferuje trzy rodzaje wniosków, a mianowicie specjalny 510(k), skrócony 510(k) i tradycyjny 510(k). W 2019 r. US FDA wydała specjalny dokument zawierający wytyczne dotyczące 510(k), opisujący opcjonalną ścieżkę dla producentów, którzy wprowadzają określone, dobrze zdefiniowane modyfikacje do swoich legalnie wprowadzonych do obrotu urządzeń.
Dlaczego specjalny 510(k)?
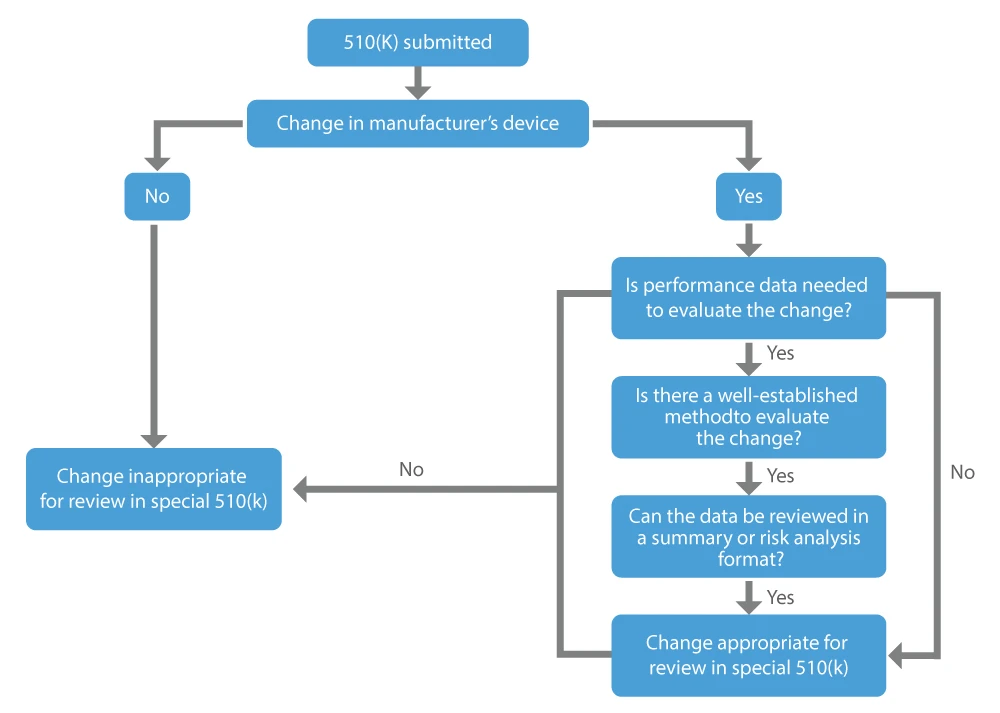
Gdy producent chce uzyskać zgodę na modyfikacje wprowadzone w już wprowadzonym do obrotu urządzeniu, tj. istniejącym urządzeniu, może złożyć wniosek o specjalną 510(k). Główne czynniki, które należy wziąć pod uwagę przy określaniu, czy zmiana w istniejącym urządzeniu może być odpowiednia dla specjalnego 510(k), są następujące:
- Zmiana dotyczy własnego, legalnie wprowadzonego do obrotu urządzenia predykcyjnego.
- Dane dotyczące wydajności nie są wymagane lub dostępne są ugruntowane metody, jeśli zostaną uznane za niezbędne do oceny zmiany.
- Wszystkie dane dotyczące wydajności na poparcie stwierdzenia istotnej równoważności mogą zostać poddane przeglądowi w formie podsumowania lub analizy ryzyka.
Dokumenty wymagane dla specjalnego 510(k)
- List motywacyjny
- Nazwa legalnie wprowadzonego do obrotu (istniejącego) urządzenia producenta i numer 510(k)
- Szczegółowy opis zmian wprowadzonych w urządzeniu, które spowodowały złożenie nowego wniosku 510(k)
- Porównanie zmodyfikowanego urządzenia z oczyszczonym urządzeniem w formacie tabelarycznym
- Inne zmiany w oznakowaniu lub projekcie
- Zwięzłe podsumowanie działań związanych z kontrolą projektu
- W oparciu o analizę ryzyka, identyfikacja działań weryfikacyjnych i/lub walidacyjnych wymaganych do zachowania zgodności z 21 CFR 820.30
- Formularz wskazań do stosowania
- Oświadczenie, że podmiot przekazujący dane spełnił i obecnie nie narusza wymogów procedury kontroli projektu określonych w 21 CFR 820.30, a dokumentacja jest dostępna do wglądu na żądanie.
Specjalny harmonogram przeglądu 510(k) przez US agencjęFDA
Zgodnie z wytycznymi FDA"Refuse to Accept Policy for 510(k)s", termin przeglądu specjalnych zgłoszeń 510(k) wynosi trzydzieści (30) dni od ich otrzymania.
Kiedy ubiegać się o specjalny 510(k)?
US FDA nieustannieFDA do zapewnienia bezpiecznych i skutecznych wyroby medyczne promowania zdrowia ludzkiego. Specjalny program 510(k) jest skuteczny i zgodny z najmniej uciążliwą procedurą przeglądu, która pomaga zagranicznym producentom sprzedawać swoje urządzenia w Stanach Zjednoczonych i umożliwia pacjentom szybki dostęp do nowych wyroby medyczne.
Aby uzyskać dalsze wyjaśnienia dotyczące specjalnego procesu 510(k) FDA, reach się z Freyr - sprawdzonym ekspertem ds. regulacji. Bądź na bieżąco. Zgodność z przepisami.