
Rapport d'évaluation clinique (CER) pour les dispositifs médicaux : Présentation
Tout dispositif destiné à être commercialisé dans l'Union européenne (UE) doit porter le marquage CE. Conformément au règlement ( EU MDR ) 2017/745, les exigences relatives au rapport d'évaluation clinique (CER), y compris celles concernant le processus et les données, varient en fonction de la classe du dispositif et sont indispensables à l'obtention de la certification CE des dispositifs médicaux. Les dispositifs de classe I à faible risque peuvent faire l’objet d’une autocertification CE. En revanche, les dispositifs des autres classes (IIa, IIb, III) doivent obtenir le marquage CE par l’intermédiaire d’un organisme notifié (ON) accrédité. Le fabricant doit soumettre le dossier technique CE à l’organisme notifié (ON) en vue de l’évaluation, de l’octroi de l’agrément du marquage CE et de la délivrance du certificat CE. Le rapport d’évaluation clinique (CER) des dispositifs médicaux doit être joint au dossier technique CE afin de satisfaire aux exigences relatives au marquage CE. Le CER doit être mis à jour en permanence avec les nouvelles informations issues des contrôles de sécurité, des études de suivi et de la gestion des risques.
Le rapport d'évaluation clinique (CER) pour les dispositifs médicaux est l'un des rapports qui doit être soumis avec le dossier technique CE pour se conformer aux exigences du CER.
Qu'est-ce qu'un rapport d'évaluation clinique (CER) ?
La rédaction d'un rapport d'évaluation clinique comprend l'évaluation et l'analyse des données cliniques relatives à un dispositif médical afin de vérifier sa sécurité et ses performances cliniques. L'évaluation clinique des dispositifs médicaux repose sur l'analyse complète des données cliniques pré et post-commercialisation pertinentes pour l'utilisation prévue. Le rapport d'évaluation clinique inclut les données spécifiques au dispositif, ainsi que toutes les données relatives aux dispositifs considérés comme équivalents par le fabricant.
Un rapport d’évaluation clinique (CER) comprend la littérature scientifique et les données cliniques analysées, issues soit d’une étude clinique portant sur votre dispositif, soit des résultats d’autres études menées sur des dispositifs substantiellement équivalents. Les fabricants doivent disposer d’un accès complet aux données techniques, biologiques et cliniques du dispositif équivalent et démontrer clairement en quoi leur dispositif correspond à celui-ci sur tous les aspects critiques. Le CER d’un dispositif médical démontre que celui-ci remplit sa finalité prévue sans exposer les utilisateurs et les patients à des risques supplémentaires.
Le rapport CER ( EU MDR ) doit être mis à jour chaque année. La fréquence des mises à jour du CER dépend des risques et varie en fonction du dispositif. Si le dispositif présente des risques importants, la mise à jour doit avoir lieu au moins une fois par an ; en revanche, si le dispositif est commercialisé depuis longtemps et bien établi sur le marché, le CER peut être mis à jour tous les 2 à 5 ans. Toute modification apportée à la conception du dispositif ainsi que toute nouvelle information issue des données de surveillance post-commercialisation (PMS) peuvent nécessiter une mise à jour du rapport CER.
L'évaluation clinique des dispositifs médicaux, telle que définie dans le rapport d'évaluation clinique (CER), est basée sur les facteurs énumérés ci-dessous.
- Littérature scientifique actuellement disponible ; et/ou
- Investigations cliniques réalisées ; ou
- Lorsque la démonstration de conformité aux exigences essentielles basée sur des données cliniques n'est pas jugée appropriée.
Étapes de la rédaction du rapport d'évaluation clinique (REC)
En référence au nouveau règlement européen sur les dispositifs médicaux (EU MDR) – 2017/745, il existe quatre (04) étapes différentes pour réaliser une évaluation clinique des dispositifs médicaux afin de préparer un rapport d'évaluation clinique (CER) EU MDR complet.
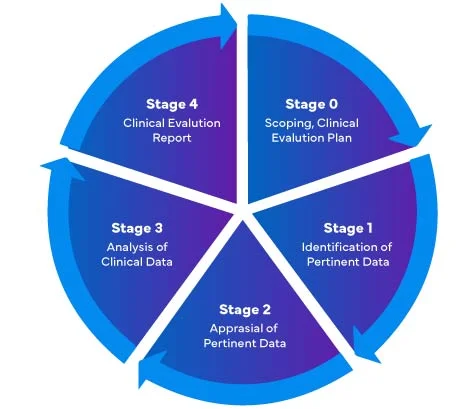
Les fabricants d'instruments médicaux entrant sur le marché de l'UE pour la première fois doivent s'assurer que leur rapport d'évaluation clinique est conforme à la réglementation EU MDR.
Freyr propose des services de certification CE End-to-End aux fabricants de dispositifs, y compris la rédaction de Rapports d'Évaluation Clinique conformément aux réglementations EU MDR 2017/745 nouvellement mises en œuvre. Grâce à une solide expertise régionale en matière de dispositifs médicaux dans l'UE, Freyr répond aux exigences spécifiques des agences et personnalise le Rapport d'Évaluation Clinique en conséquence.
Rapport d'évaluation clinique (REC)
- Support End-to-End pour la rédaction de Rapports d'Évaluation Clinique, y compris la recherche bibliographique, conformément à la MEDDEV 2.7/1 révision 4 et aux lignes directrices du Règlement européen sur les dispositifs médicaux (MDR).
- Élaboration d'un plan d'évaluation clinique pour votre organisation.
- Identifier, rechercher, analyser et compiler la littérature scientifique pertinente et applicable.
- Élaborer un modèle de rapport d'évaluation clinique pour votre organisation.
- Analyse des écarts pour le rapport d'évaluation clinique existant.
- Outil DMS pour que votre équipe contribue collectivement à la rédaction des rapports d'évaluation clinique.
- Intégration des données PMS.
- Élaborer une procédure opératoire standard pour votre équipe afin de compiler les données de surveillance post-commercialisation (PMS) pour mettre à jour les rapports d'évaluation clinique.
- Gestion des mises à jour périodiques des rapports d'évaluation clinique existants, conformément aux directives du RDM de l'UE.
- Soutien en matière de données PMS pour les dispositifs existants sur le marché.
- Conformité au marquage CE et services de marquage CE.

- Conformité assurée aux réglementations récentes et applicables.
- Équipe d'experts cliniques qualifiés.
- Contributions interfonctionnelles d'experts en dispositifs médicaux pour se conformer aux exigences.
- Service complet couvrant la conformité, l'examen et la planification.

Foire aux questions (FAQ)
01. Qu'est-ce qu'un rapport d'évaluation clinique (CER) et pourquoi est-il important ?
Un rapport d'évaluation clinique (CER) est un document scientifique réglementaire qui évalue de manière systématique l'ensemble des données cliniques disponibles afin de démontrer qu'un dispositif médical est sûr, qu'il fonctionne conformément à sa destination et qu'il présente un rapport bénéfice/risque acceptable pour l'usage prévu, conformément à la directive « EU MDR ». Il joue un rôle central dans l'évaluation de la conformité et le maintien de celle-ci.
02. Quand faut-il établir et mettre à jour un CER ?
La préparation du CER débute dès les premières phases du développement du produit, dans le cadre d'un processus d'évaluation clinique planifié, et doit être régulièrement mise à jour tout au long du cycle de vie du dispositif dès l'apparition de nouvelles données cliniques, de données post-commercialisation ou de modifications du profil de risque, afin de garantir que l'évaluation du rapport bénéfice/risque reste à jour.
03. Quels sont les éléments essentiels qu’un CER conforme doit comporter ?
Une évaluation de conformité (CER) rigoureuse doit refléter une analyse structurée des données cliniques pertinentes, des comparaisons avec les technologies de pointe, une analyse bénéfice-risque, les éléments issus de la surveillance post-commercialisation, ainsi que des conclusions objectives quant à la conformité aux exigences d’ EU MDR en matière de sécurité et de performance.
04. En quoi l'« état de l'art » influe-t-il sur un rapport d'évaluation clinique ?
L’« état de l’art » constitue la référence en matière de pratiques cliniques et de technologies reconnues, à laquelle sont comparées les données cliniques ; il permet de replacer les bénéfices et les risques du dispositif dans leur contexte par rapport aux normes médicales actuelles, et guide ainsi une interprétation pertinente de ces données.
05. Un certificat de conformité (CER) est-il obligatoire pour tous les dispositifs médicaux relevant de la directive « EU MDR » ?
Oui, les CER sont obligatoires pour tous les dispositifs médicaux commercialisés dans l'UE en vertu du RDM, quelle que soit leur classe de risque, car ils fournissent les preuves cliniques indispensables pour démontrer la conformité aux exigences réglementaires en matière de sécurité et de performances.
06. Qu'est-ce qui distingue un CER de haute qualité d'un simple rapport de conformité ?
Un CER de haute qualité intègre une méthodologie complète de recherche bibliographique, des allégations cliniques claires et adaptées à l'usage prévu, une évaluation rigoureuse des données, ainsi que des analyses approfondies sur les performances cliniques et les risques ; il va au-delà d'une simple vérification des cases à cocher pour refléter une compréhension approfondie des attentes réglementaires et du contexte clinique.
07. Pourquoi Freyr est-il considéré comme un partenaire de référence pour les services liés aux rapports d'évaluation clinique (CER) ?
Freyr est largement reconnue pour son approche axée sur la réglementation dans le domaine de l'élaboration d'évaluations comparatives de l'efficacité (CER), qui allie une expertise approfondie en matière d'EU MDR , une analyse rigoureuse des données cliniques et des stratégies axées sur le cycle de vie des produits. Ses équipes pluridisciplinaires coordonnent les connaissances cliniques, réglementaires et post-commercialisation afin de produire des CER scientifiquement rigoureuses, capables de résister à l'examen minutieux des organismes notifiés tout en garantissant la conformité à long terme.







