Présentation des produits combinés de dispositifs médicaux
Dans le monde dynamique des soins de santé et de l'innovation, les produits combinés de dispositifs médicaux sont devenus un pont solide reliant les produits pharmaceutiques, les dispositifs médicaux et les produits biologiques. Le marché des produits combinés est en pleine croissance, avec un taux de croissance annuel composé (TCAC) anticipé de 8,9 % entre 2023 et 2030. Le secteur des produits combinés médicaments-dispositifs est promis à une croissance soutenue, soutenu par les avancées technologiques, l'amélioration des infrastructures de santé, des voies réglementaires plus fluides, des collaborations stratégiques et un engagement envers des soins centrés sur le patient.
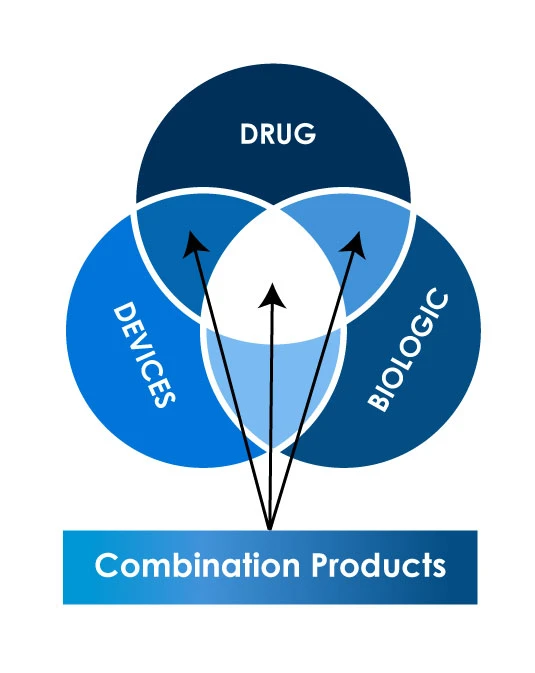
Différents types de produits combinés
Scénario réglementaire mondial pour l'enregistrement des produits combinés
L'interprétation de ce qui constitue un produit combiné peut différer d'un pays à l'autre, ce qui complique l'enregistrement de ces produits dans diverses nations. De plus, les exigences et procédures réglementaires pour les produits combinés peuvent présenter des variations en matière de documentation, de communication et de validation. Le paysage réglementaire pour l'enregistrement des produits combinés peut différer considérablement à l'échelle mondiale. Voici les principales Autorités Réglementaires qui supervisent ces dispositifs au niveau mondial.
| Pays | Agence | Centres principaux d'approbation |
|---|---|---|
| ÉTATS-UNIS | Bureau des produits combinés (OCP) | Centre d'évaluation et de recherche des médicaments (CDER) |
| Centre d'évaluation et de recherche des produits biologiques (CBER) | ||
| Centre des dispositifs médicaux et de la santé radiologique (CDRH) | ||
| UE | Organismes Notifiés (ON) | Autorité nationale compétente (Produits médicaux) |
| Organismes Notifiés (ON) (dispositifs médicaux) | ||
| Japon | Division de l'évaluation et des licences ou le Bureau des dispositifs médicaux / produits à base de cellules et de tissus du Bureau de la sécurité pharmaceutique et alimentaire | Directeur de la Division de l'évaluation et de l'autorisation (DMDL), Bureau de la sécurité pharmaceutique et alimentaire, Bureau de la sécurité pharmaceutique et médicale, Ministère de la Santé et du Bien-être |
| Chine | Center for Medical Device Standardization Administration (CMDSA) | Centre d'évaluation des dispositifs médicaux (CMDE) |
| Center of Drug Evaluation (CDE) | ||
| Malaisie | Agence Nationale de Réglementation Pharmaceutique | Agence nationale de réglementation pharmaceutique (NPRA) |
| Agence des dispositifs médicaux |
L'enregistrement des produits combinés sur les marchés internationaux nécessite une approche personnalisée, impliquant une collaboration étroite avec les agences de santé compétentes pour l'approbation. Le processus typique d'enregistrement des produits combinés comprend les étapes suivantes :
- Évaluer si un dispositif spécifique répond aux critères de classification en tant que produit combiné.
- Classification des dispositifs en fonction des risques associés.
- Identification des normes pertinentes et des prérequis en matière de données spécifiés par l'Agence de Santé concernée.
- Produire les données nécessaires, tel que mandaté par l'Agence.
- Compilation d'un dossier technique conformément aux exigences spécifiques de chaque pays.
- Soumettre la demande et répondre à toute question ou préoccupation jusqu'à l'obtention de l'approbation.
- Gestion du cycle de vie du dispositif après approbation.
Nos compétences
- Analyse des risques initiale
- Étude de marché - Analyses de marché spécifiques aux produits
- Renforcement des équipes
- Élaboration de la stratégie réglementaire
- Marchés potentiels et voies d'accès
- Dossier de conception et analyse des risques
- Système de Gestion de la Qualité (SGQ) ISO 13485
- Programme d'audit unique des dispositifs médicaux (MDSAP)
- Pré-évaluation QMS ISO 13485
- Stratégie réglementaire
- Freyr IMPACT (Plateforme d'Intelligence Réglementaire)
- Vérification et validation de la conception
- Gestion des risques
- Rédaction de la documentation technique
- Stratégie réglementaire
- Exigences réglementaires
- L'outil rDMS de Freyr (Système de gestion des données/de la documentation)
- Validation des processus et validation clinique.
- Étiquetage et Artwork finaux
- Représentation locale
- Dépôt réglementaire
- Le marquage « Conformité Européenne » (CE) de l'Union européenne (UE) et le marquage UK Conformity Assessment (UKCA)
- Certification d'accès au marché mondial
- Soutien à l'audit par l'organisme notifié (ON)/organisme agréé
- Représentation locale
- Approbations réglementaires
- Surveillance après commercialisation (PMS)
- Suivi clinique après commercialisation (PMCF)
- Maintenance annuelle du dossier technique (CER/gestion des risques)
- Renouvellements réglementaires
- Nouveaux lancements sur le marché
- Communication avec l'Autorité compétente/l'Organisme notifié/l'Organisme agréé
- Solutions de pharmacovigilance (PV) automatisées
Pourquoi Freyr ?
Enregistrement des dispositifs médicaux
- Stratégie réglementaire complète pour les produits combinés.
- Soutien réglementaire pour les documents de développement de produits tels que les dossiers historiques de conception (DHFs).
- Stratégie de conformité SMQ.
- Conformité réglementaire, analyse des écarts et remédiation des documents techniques et des systèmes qualité.
- Services d'étiquetage réglementaire et de rédaction technique.
- Services de veille réglementaire et de marché.
- Services de traduction de documents et d'étiquetage.
- Liaison et service avec l'Agence de santé.
- Services d'Artwork réglementaires.
- Services de pharmacovigilance et de PMS.
- Services de publication.
- Services de rédaction médicale.

- Soumissions réussies pour diverses classes de DIV.
- Personnel dédié et expert pour fournir un soutien réglementaire aux dispositifs médicaux et aux DIV.
- Soumission dans les délais des livrables.
- Accès à une filiale locale pour relever les défis de l'Autorité et les exigences linguistiques spécifiques.
- Assistance d'un représentant local ou légal avec un modèle économique.
- Services de gestion des ressources réglementaires / de renforcement des équipes.