
Aperçu des services de conformité IVDR de l'UE
Le règlement européen relatif aux dispositifs médicaux de diagnostic in vitro (EU IVDR) est une nouvelle base réglementaire pour la mise sur le marché européen des dispositifs de diagnostic in vitro (DDIV). Le règlement EU IVDR est destiné à remplacer la directive actuelle de l'UE relative aux dispositifs médicaux de diagnostic in vitro (UE 98/79/CE). En tant que règlement européen, il sera applicable dans tous les Member States de l'UE et les États de l'Association européenne de libre-échange (AELE) sans qu'il soit nécessaire de le transposer dans le droit des États respectifs. En tant que partenaire réglementaire éprouvé, Freyr fournit des services de conformité EU IVDR et des services de conseil IVDR aux fabricants pour des réglementations de diagnostic in vitro conformes et le marquage CE.
Le Règlement IVDR - Calendrier de transition
Entré en vigueur le 26 mai 2022, le nouveau règlement européen sur les DM DIV (IVDR) est considéré comme un changement significatif dans l'encadrement réglementaire des DM DIV. Un aperçu de la transition IVDR est présenté ci-dessous –
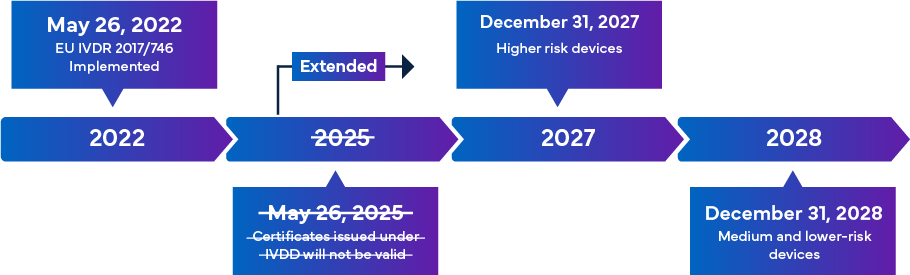
Une fois pleinement mises en œuvre, les règles de conformité à l'IVDR garantissent que presque tous les DIV entrant sur le marché européen sont soumis à l'examen de l'organisme notifié (ON) de l'IVDR et à la certification de marquage CE dans le cadre du processus d'approbation des DIV. Dans ce scénario, outre l'alignement avec la classification IVDR mise à jour, il est impératif pour les fabricants d'examiner efficacement la documentation technique clé pour un enregistrement et un marquage CE réussis des DIV. Les exigences de l'IVDR varient en fonction de la classe de risque du DIV. En général, pour la certification des DIV, les fabricants doivent effectuer :
- Examens des dossiers techniques conformément au règlement IVDR (Règlement UE 2017/746)
- Préparation des Rapports d'évaluation des performances (PER) pour tous les DIV
- Suivi des performances après commercialisation (PMPF) conformément à l'annexe XIII partie B de l'IVDR
- Déclaration de vigilance conformément à l'article 82 de l'IVDR
Freyr Solutions aide ses clients à réaliser une revue systématique de la littérature scientifique et à la planification et à la préparation d'un PER pour la conformité au Règlement sur les dispositifs médicaux de diagnostic in vitro. Freyr Solutions dispose d'experts dédiés qui fournissent des services de conseil IVDR et un soutien en matière de surveillance après commercialisation (PMS), qui fait partie intégrante du système de gestion de la qualité du fabricant, ainsi que du suivi des performances après commercialisation (PMPF).
Obtenez des conseils d'experts sur votre conformité au règlement européen IVDR
Expertise et avantages des services de conformité IVDR de l'UE
- Plan de transition pour la conformité au IVDR
- Examen technique et analyse des écarts des exigences de l'IVDR pour les GSPR (Exigences générales en matière de sécurité et de performances)
- Soutien à la compilation du dossier technique conformément aux exigences de l'IVDR
- Rapports de validité scientifique basés sur la littérature et/ou des données internes
- Rapports de performance clinique basés sur la littérature et/ou des données internes
- Preuves cliniques ou rapports d'évaluation des performances
- Protocoles et rapports de suivi des performances après commercialisation (PMPF)
- Protocoles et rapports de surveillance après commercialisation (PMS)
- Rédaction/révision d'autres documents tels que la notice d'emballage/IFU (Instructions d'utilisation), les Instructions de référence rapide (QRI), le Manuel d'utilisation/d'opération, etc.

- Conformité IVDR assurée, enregistrement des DIV et marquage CE
- Solide compréhension réglementaire et expertise dans les domaines clés d'impact du règlement européen IVDR
- Modèle de livraison axé sur une gestion de projet solide pour garantir le respect des délais
- Experts NB internes (examen du rapport par les examinateurs interactifs NB)
- Équipes dédiées avec une expertise transversale sur des domaines d'impact spécifiques et des catégories de dispositifs
- Contributions interfonctionnelles d'experts en dispositifs médicaux pour se conformer aux exigences
- Gamme complète de services couvrant la conformité, l'examen et la planification
- Solide expertise pour assurer la cohérence des livrables (délai et qualité)








