
Conformité en matière d'enregistrement des dispositifs médicaux et cadre réglementaire dans l'Union européenne
L’Union européenne représente l’un des marchés des dispositifs médicaux les plus réglementés et les plus attractifs au monde sur le plan commercial. Avec les 27 member states, les 3 EEA pays et la Turquie, le marché de l’Union fonctionne selon un cadre réglementaire harmonisé défini par le règlement EU MDR (2017/745) pour les dispositifs médicaux et le règlement IVDR de l’UE (2017/746) pour les dispositifs de diagnostic in vitro ; la région garantit ainsi des normes uniformes de sécurité et de performance pour toutes les catégories de produits. Pour commercialiser légalement un dispositif sur le marché de l’Union européenne, les fabricants doivent obtenir le marquage CE, qui atteste de la conformité à la réglementation applicable en fonction de la classification des risques du dispositif et de la voie d’évaluation de la conformité. Le non-respect de ces exigences peut entraîner de graves conséquences, notamment le rappel du produit, sa saisie à la douane, la suspension de la certification ou la perte totale de l’accès au marché.
Dans le cadre du régime réglementaire actuel :
- Le marquage « CE » est obligatoire pour tous les dispositifs médicaux et les dispositifs de diagnostic in vitro avant leur mise sur le marché.
- L'enregistrement des dispositifs médicaux dans EUDAMED est mis en place par étapes, et plusieurs modules sont déjà opérationnels et leur utilisation est obligatoire.
- La désignation d'un représentant autorisé européen (EAR) est obligatoire pour tout fabricant établi en dehors de l'UE, de l'EEA e ou de la Turquie.
- Les règles de classification plus strictes prévues par les règlements MDR et IVDR exigent une documentation technique solide, des données cliniques ou des preuves de performance, ainsi qu'un suivi continu tout au long du cycle de vie des dispositifs.
Freyr accompagne les fabricants tout au long de ce parcours en leur permettant d'accéder au marché de l'Union européenne en toute fluidité et dans le respect de la réglementation. De l'élaboration d'une stratégie de marquage CE et de la constitution du dossier technique à la gestion des demandes auprès des organismes notifiés et à la fonction de représentant autorisé dans l'UE, Freyr garantit une conformité totale aux règlements MDR et IVDR ainsi qu'un accès sans entrave à l'ensemble des régions de l'UE member states, EEA ainsi qu'à la Turquie.
Processus de mise en conformité avec la réglementation européenne, étape par étape
La mise sur le marché d'un dispositif médical ou d'un dispositif de diagnostic in vitro (IVD) au sein de l'Union européenne (UE) comporte plusieurs étapes bien définies. Voici comment Freyr gère l'ensemble du processus pour vous :
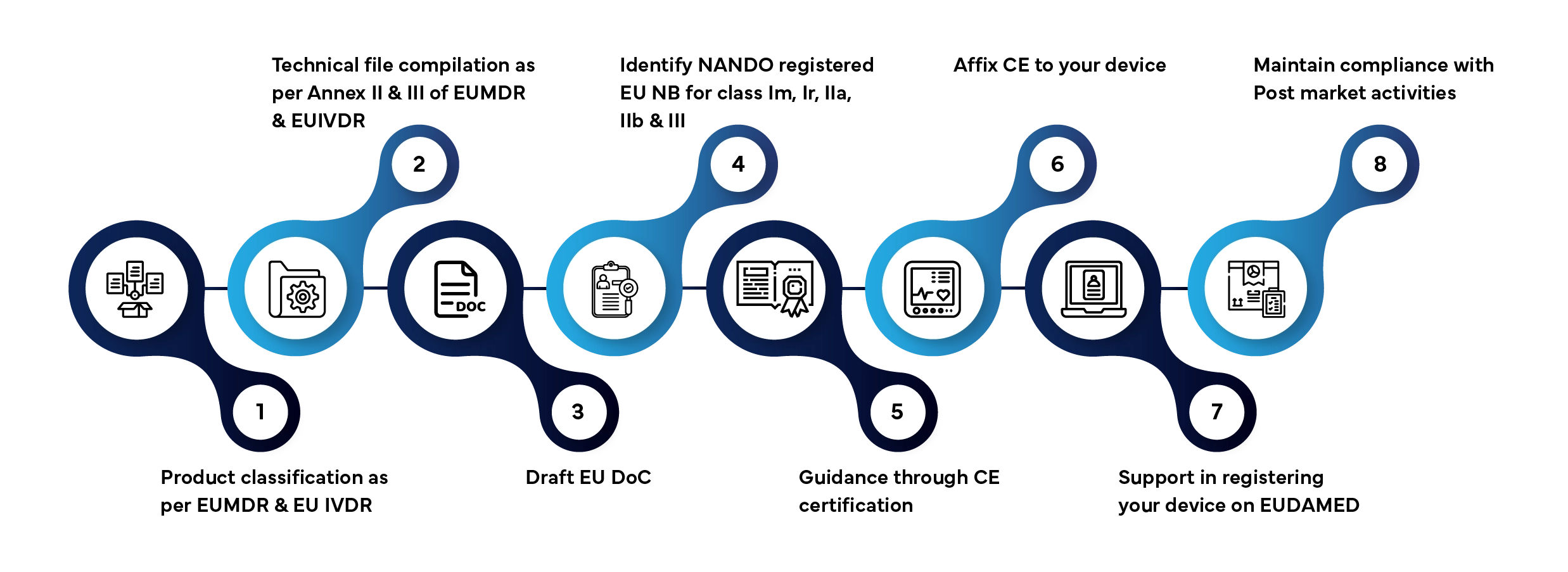
Délai de traitement habituel : 3 à 12 mois, en fonction de la catégorie du dispositif et de l'état d'avancement du dossier.
Principales offres de Freyr Medical Device dans l'Union européenne
- Stratégie réglementaire et transition vers le MDR et l'IVDR – Nous élaborons des feuilles de route réglementaires end-to-end adaptées à l'EU MDR e et à l'IVDR de l'UE, afin de garantir une transition en douceur depuis les régimes régis par les directives et une mise en conformité avec les exigences actuelles de l'UE.
- Documentation technique et évaluation de la conformité – Notre équipe vous accompagne dans l'élaboration, la révision et le dépôt de vos dossiers techniques / dossiers de conception, vous aide dans vos démarches auprès des organismes notifiés et gère le marquage « CE », les essais de sécurité et de performance des dispositifs médicaux, ainsi que les procédures de conformité.
- Évaluation clinique et de performance : Freyr assure la rédaction par des experts de rapports d’évaluation clinique (CER), de rapports d’évaluation de performance (PER), de plans de surveillance post-commercialisation ( PMCF) et de plans de surveillance post-commercialisation des dispositifs médicaux (PMPF), de rapports de sécurité post-commercialisation (PSUR), de rapports de survie virale (SVR), de rapports de performance clinique (CPR) pour les dispositifs de diagnostic in vitro (IVD), de documentation relative aux risques et de contenus d’évaluation biologique, garantissant ainsi la clarté technique et la conformité réglementaire pour toutes les classes de dispositifs.
- Assistance à l'enregistrement UDI et EUDAMED – Nous veillons à la conformité de votre système d'identification unique des dispositifs médicaux (UDI) et vous accompagnons dans votre enregistrement auprès de la base de données européenne des dispositifs médicaux EUDAMED, ainsi que dans la gestion du cycle de vie associée.
- Représentant autorisé européen (EAR) et représentation locale – Pour les fabricants situés en dehors de l'UE, de l'EEA e et de la Turquie, nous agissons en tant que votre représentant autorisé européen (EAR) mandaté et vous fournissons une assistance locale en matière de conformité dans toute l'UE member states.
- Surveillance post-commercialisation (PMS) – Freyr vous accompagne dans la mise en place, la mise en œuvre et le maintien de systèmes de surveillance post-commercialisation, notamment le plan de surveillance post-commercialisation (PMSP), le rapport de surveillance post-commercialisation (PMSR), le rapport périodique de mise à jour sur la sécurité (PSUR), les plans de suivi clinique et de performance post-commercialisation (PMCF/PMPF), les déclarations de vigilance, les FSCAs, les FSNs et les CAPAs, garantissant ainsi le maintien continu du marquage CE et un accès durable au marché, dans le respect de la conformité tout au long du cycle de vie du produit.
- Enregistrement des dispositifs médicaux au titre du marquage CE : Freyr prend en charge l'ensemble du processus d'enregistrement au titre du marquage CE dans l'UE en accompagnant l'évaluation de la conformité, en constituant les dossiers de conformité, en collaborant avec les organismes notifiés et en garantissant l'obtention des autorisations dans les délais impartis pour toutes les catégories de dispositifs médicaux et de dispositifs de diagnostic in vitro.
- Accompagnement en matière de SMQ : Nous accompagnons la mise en place et le maintien de systèmes de gestion de la qualité conformes à la norme « ISO 13485 », en adéquation avec les exigences de qualité et de sécurité du règlement « EU MDR » / IVDR ainsi qu’avec les attentes des organismes notifiés.
- Conformité du labelling:Notre équipe veille à ce que vos étiquettes, notices d'utilisation, emballages et symboles soient conformes aux exigences du règlement MDR/IVDR et aux règles linguistiques multilingues de l'UE, en garantissant la cohérence et la conformité dans les 27 États membres de l'UE member states.
Offres de services du représentant agréé de l'UE (EAR)

Enregistrement des dispositifs auprès des autorités de l'UE
Pour les fabricants hors UE, Freyr agit en tant que représentant autorisé désigné (EAR) au sein de l'UE, conformément aux exigences du RDM et de la DIVM. En tant qu'entité établie en Allemagne, Freyr accompagne les activités d'enregistrement des dispositifs auprès de l'autorité compétente allemande concernée et tient à jour les dossiers réglementaires requis. Pour les autres Member States de l'UE, Freyr fournit un accompagnement réglementaire, le cas échéant, afin de garantir la conformité des dispositifs et de faciliter leur mise sur le marché de l'Union.

Documentation et assurance de la conformité
Nos experts en réglementation vérifient que votre déclaration de conformité (DoC), vos certificats CE et vos dossiers techniques sont complets, à jour et conformes aux règlements MDR et IVDR. Freyr garantit que tout est prêt pour l'évaluation de la conformité et le dépôt des demandes de marquage CE.

Réponse aux demandes des autorités compétentes
Si nécessaire, Freyr gère pour votre compte toutes les communications directes et les demandes de précisions émanant des autorités compétentes de l'UE ou des organismes notifiés, en veillant à fournir des réponses précises et dans les délais, et en contribuant ainsi à éviter tout retard dans les procédures d'autorisation ou les contrôles réglementaires post-commercialisation.

Vigilance et communication en cas d'incident
En tant que responsable de la sécurité des dispositifs médicaux (EAR), Freyr assure la liaison principale pour toutes les communications relatives à la sécurité. Si nécessaire, nous coordonnons les notifications d'incidents, les mesures correctives de sécurité sur le terrain (FSCA) et les rapports de vigilance entre le fabricant, les professionnels de santé et les autorités, afin de garantir une réponse adéquate et le respect de la réglementation.

Préparation aux inspections et aux audits
Freyr conserve l'ensemble de la documentation, de la correspondance et des registres obligatoires en vue des audits et inspections menés par les autorités. Notre équipe veille à ce que la documentation technique, l'étiquetage et les registres post-commercialisation soient facilement accessibles et pleinement conformes aux exigences du MDR et de l'IVDR.
Pourquoi collaborer avec Freyr ?
- End-to-end une expertise réglementaire qui couvre l'ensemble du processus, de l'enregistrement préalable à la mise sur le marché à la vigilance post-commercialisation, en gérant chaque étape de la mise en conformité.
- Une expérience éprouvée, avec plus de 2 000 enregistrements d'appareils menés à bien dans diverses catégories.
- Une forte présence européenne implantée en Allemagne (EAR), complétée par des spécialistes de la réglementation sur place à Reading et soutenue par les équipes opérationnelles internationales de Freyr.
- Une planification sur mesure de la transition, offrant un accompagnement stratégique pour obtenir la certification CE en toute sérénité et à moindre coût.
- Des équipes de livraison multirégionales bénéficiant d'un soutien sur le terrain grâce aux activités européennes de Freyr, basées en Allemagne.
- Expérience avérée en matière de mise en conformité avec les règlements MDR et IVDR et de transition des dispositifs existants
- Une gouvernance transparente du projet grâce à une communication directe avec l'organisme notifié

Célébrer le succès de nos clients
Dispositifs médicaux
Assistance à l'enregistrement et LR
Mondial
Freyr a été un partenaire indispensable pour atteindre une évolutivité mondiale rapide pour notre activité de Software as a Medical Device (SaMD). En tant que startup, l'acquisition d'une expertise en matière de réglementations mondiales est prohibitive en termes de coûts. La tarification compétitive et les services personnalisés de Freyr nous ont permis d'obtenir cette expertise à une fraction du coût des ressources à temps plein. La réactivité et l'adaptabilité de leur équipe aux priorités des projets ont grandement facilité nos progrès. Nous recommandons Freyr à toute entreprise recherchant des conseils et un soutien experts dans le domaine réglementaire des dispositifs médicaux.
Arie Henkin
VP - Qualité et Réglementation, basée en Australie, entreprise leader en SaMD.
Dispositifs médicaux
Services de représentant suisse
Japon et Suisse
J'apprécie vraiment ma collaboration avec Freyr, et je les considère comme un atout précieux et une extension de ma propre équipe. Ils sont fiables et précis, et leurs tarifs sont compétitifs. De plus, je n'hésiterai pas à collaborer à nouveau avec Freyr.
Darren Mansell
Responsable des affaires réglementaires, basé au Royaume-Uni, pour une entreprise mondiale de conception et de fabrication de dispositifs médicaux
Dispositifs médicaux
Services d'enregistrement et AR
Malaisie et Indonésie
Freyr offre un service fiable avec une expertise dans de nombreux pays. Je peux compter sur Freyr pour fournir les informations nécessaires à une décision éclairée avant de conclure un accord formel sur l'étendue des travaux. Une fois qu'un projet est lancé, l'équipe Freyr agit de manière professionnelle pour exécuter le travail avec une excellente communication sur l'avancement.







