Aperçu de l'enregistrement des dispositifs médicaux auprès de la US FDA.
Les États-Unis d'Amérique (USA) sont réputés pour être un marché très réglementé pour les dispositifs médicaux, avec des voies et des exigences d'enregistrement bien définies. Les premières réglementations américaines sur les dispositifs médicaux remontent à 1976 et ont évolué au fil du temps. Elles sont réglementées par le Centre for Devices and Radiological Health (CDRH) sous l'égide de la Food and Drug Administration (FDA). Freyr a aidé de nombreux fabricants de dispositifs à se conformer au processus d'enregistrement des dispositifs médicaux de la US FDA.
![]()
Autorité réglementaire : Food and Drug Administration (FDA)![]()
Réglementation : Titre 21 du Code des règlements fédéraux (21 CFR) Parties 800 – 1299![]()
Voie réglementaire : Notification préalable à la commercialisation ou Autorisation préalable à la commercialisation ou Classification De Novo![]()
Représentant autorisé : Agent US![]()
Exigence SMQ : Réglementation du Système Qualité (QSR) (21 CFR partie 820)![]()
Évaluation des données techniques : Centre pour les dispositifs et la santé radiologique![]()
Validité de la licence : Illimité![]()
Exigences d'étiquetage : 21 CFR Partie 801![]()
Format de soumission : Papier et CD/DVD![]()
Langue : Anglais
Classification des dispositifs médicaux aux ÉTATS-UNIS
La FDA classe les dispositifs médicaux en 3 catégories basées sur le risque : Classe I, Classe II et Classe III. Les dispositifs de Classe I sont considérés comme à faible risque, tandis que ceux de Classe III sont associés à un risque élevé. Les exigences d'enregistrement et le processus varient en fonction de la classe du dispositif.
| Classe de dispositif | Risque | Voie d'enregistrement pour approbation |
|---|---|---|
| I | Faible risque | Exempté 510(k) |
| II | Risque modéré (Avec dispositif de référence) | Notification précommercialisation / 510(k) |
Risque modéré (Sans dispositif de référence) | Demande De Novo | |
| III | Risque élevé | Approbation précommercialisation (PMA) |
Agent de la FDA US
Les entreprises sans bureaux locaux aux US doivent désigner un agent US FDA pour représenter le fabricant. L' agent US FDA doit soit résider aux US, soit y maintenir un lieu d'affaires. Les responsabilités à remplir par l'agent sont prédéterminées par la US FDA dans le cadre des réglementations CFR.
Parcourez la Foire aux questions (FAQ) concernant l'Agent US.
Réunions interactives avec la FDA US
La FDA US soutient les fabricants par le biais de divers types de réunions de Q-Submission pour atteindre différents objectifs. Ces réunions avec l'agence, avant le début ou pendant le développement d'un dispositif, et avant la soumission des demandes d'enregistrement de dispositifs médicaux auprès de la FDA US, aident les fabricants à optimiser les délais et les coûts liés à la commercialisation des dispositifs.
Enregistrement des dispositifs médicaux aux ÉTATS-UNIS
Les dispositifs peuvent être approuvés par le CDRH, FDA par l'une des diverses voies d'enregistrement. Ils sont listés comme suit :
Dispositifs médicaux de classe I : Les dispositifs de classe I sont généralement exemptés des Bonnes Pratiques de Fabrication (BPF) et de la soumission 510(k) et ne nécessitent pas d'approbation préalable de la FDA US pour être commercialisés aux US. D'autres exigences, telles que l'enregistrement de l'établissement, l'inscription du dispositif, l'UDI, la PMS, etc., doivent être respectées par le fabricant.
Dispositifs médicaux de classe II : Les dispositifs à risque moyen avec des dispositifs prédicats approuvés 510(k) peuvent opter pour une Notification pré-commercialisation 510(k) (PMN), également appelée enregistrement 510(k). Le dispositif concerné doit établir une Équivalence Substantielle (ES) avec les dispositifs prédicats identifiés et revendiqués. Cette voie est la plus largement adoptée pour l'enregistrement des dispositifs aux US. Les fabricants de dispositifs à risque moyen sans prédicats peuvent demander une classification par la FDA US par le biais de demandes De-Novo.
Dispositifs médicaux de classe III : Les fabricants de dispositifs de classe III à haut risque doivent soumettre une demande d'Approbation pré-commercialisation (PMA) à la FDA US. Les dispositifs doivent faire l'objet d'une évaluation clinique détaillée et le fabricant doit soumettre des données détaillées sur la sécurité et l'efficacité issues d'études cliniques. La FDA US effectuerait une inspection du Système de Gestion de la Qualité (SGC) dans le cadre de l'évaluation avant de délivrer une Approbation pré-commercialisation pour le dispositif.
Enregistrements de dispositifs médicaux non-CDRH
Selon les indications d'utilisation, certains produits frontières considérés comme des dispositifs médicaux dans d'autres pays, tels que les respirateurs chirurgicaux, les désinfectants et les produits combinés, impliquent d'autres agences comme le Centre de contrôle des maladies (CDC), l'Institut national pour la sécurité et la santé au travail (NIOSH), l'Agence de protection de l'environnement (EPA), le Centre d'évaluation et de recherche biologique (CBER) et le Centre d'évaluation et de recherche sur les médicaments (CDER).
Exigences de conformité post-approbation pour les dispositifs médicaux
Tous les fabricants de dispositifs doivent se conformer aux exigences post-approbation énumérées ci-dessous :
- Exigence d'enregistrement et de référencement: Les établissements de toutes les classes de dispositifs doivent être enregistrés dans la base de données FURLs et le dispositif doit être référencé après l'obtention de l'approbation et avant sa commercialisation aux US. Certains dispositifs, tels que les dispositifs à rayonnement, doivent se conformer à d'autres exigences, comme un numéro d'accès, avant de pouvoir être importés aux US.
- Identification unique des dispositifs : Toutes les classes de dispositifs doivent se conformer aux réglementations relatives à l'identification unique des dispositifs (UDI) pour commercialiser les dispositifs aux US.
- Frais d'établissement: Le fabricant doit payer les frais annuels d'établissement pour maintenir l'enregistrement de son établissement actif et continuer à commercialiser des dispositifs aux US. La FDA US a une structure de frais réduite pour les petites entités disposant d'un Certificat de petite entreprise actif.
- Audits qualité: Pour les dispositifs qui ne sont pas exemptés des BPF, la FDA US peut inspecter l'établissement de fabrication à tout moment pour vérifier la conformité aux Réglementations des Systèmes Qualité (RSQ) conformément au 21 CFR 820.
Flux de processus
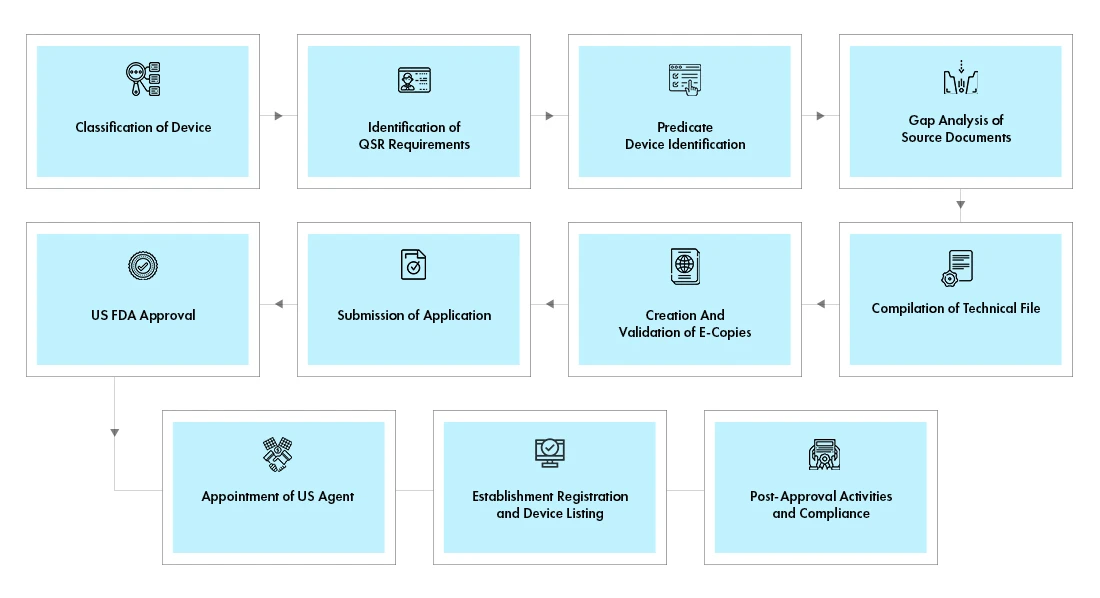
Gestion du cycle de vie des dispositifs post-approbation
Freyr accompagne les fabricants étrangers dans la gestion End-to-End du cycle de vie des dispositifs médicaux, y compris les activités post-approbation, telles que :
- Gestion des changements post-approbation – modifications aux approbations existantes de dispositifs médicaux, telles que l'ajout de nouvelles variantes, d'accessoires ; l'ajout de nouvelles indications d'utilisation, entre autres
- Maintien des approbations et de l'enregistrement grâce au paiement ponctuel des frais MDUFA à la FDA
- Assurer la liaison entre la FDA US et le fabricant
Freyr dispose d'un centre de livraison exclusif aux US avec une équipe professionnelle pour fournir un soutien réglementaire aux fabricants afin de maintenir la qualité et la sécurité nécessaires à l'approbation. Les experts en veille de Freyr observent attentivement les mises à jour réglementaires et informent les clients des mesures à prendre pour la conformité des produits avec la norme actuelle.
Résumé
| Risque | Classe de dispositif | Audit du SGC | Disponibilité du produit de référence | Voie réglementaire | US Agent | Délais de la FDA US |
|---|---|---|---|---|---|---|
| Faible risque | I | Non | NA | Exempté | Oui | 1 mois |
| Risque moyen | II | Oui (après approbation) | Oui | PMN/510(k) | Oui | 9 à 12 Mois |
| Risque moyen | II | Oui (après approbation) | Non | Demande de classification De Novo | Oui | 18 - 30 Mois |
| Risque élevé | III | Oui (avant approbation) | NA | PMA | Oui | 18 - 30 Mois |
Les services d'enregistrement de dispositifs médicaux de Freyr
L'expertise de Freyr
- Diligence raisonnable réglementaire
- Documentation des dispositifs
- Soutien 513(g)
- Enregistrement 510(k)
- Demande De Novo de classification
- Enregistrement PMA
- 21 CFR 820 conformité
- Soutien aux audits BIMO
- MDSAP Conformité
- Support d'étiquetage
- Support à la publication et à la soumission
- Agent US
- Réunions de soumission Q
- Réunions RFD et Pré-RFD
- Certification des petites entreprises
- Enregistrement de l'établissement et enregistrement des dispositifs
- Conformité réglementaire pour les dispositifs médicaux à rayonnement
- Gestion des changements post-approbation
- Surveillance après commercialisation
- Conformité UDI
- Conseil réglementaire pour la résolution des déficiences