Présentation du logiciel en tant que dispositif médical (SaMD)
Le logiciel en tant que dispositif médical (SaMD) est le sujet le plus en vogue dans le secteur de la santé. Le marché des SaMD devrait croître à un taux de croissance annuel composé (TCAC) de 10,78 % à l'échelle mondiale. Cette croissance est stimulée par divers facteurs tels que l'adoption de l'Internet des objets (IoT), les plateformes de santé numériques et l'utilisation de logiciels pour la surveillance continue des paramètres physiologiques par les professionnels de la santé pour l'assistance à distance. Cependant, ce paysage prometteur présente également des défis uniques, notamment celui de déterminer si un logiciel relève de la catégorie des dispositifs médicaux et s'il est conforme aux exigences réglementaires.
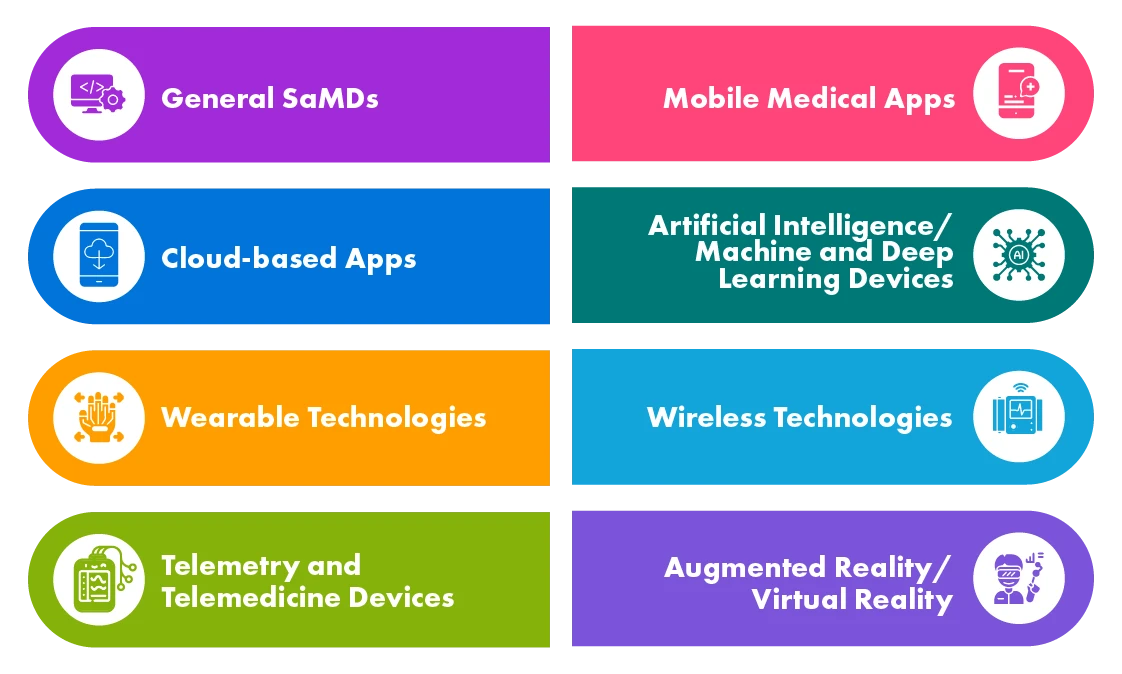
Différents types de produits de santé numériques
Scénario réglementaire mondial pour l'enregistrement des logiciels en tant que dispositifs médicaux (SaMD)
Les SaMDs sont utilisés dans diverses applications telles que le dépistage et le diagnostic, la surveillance et l'alerte, la gestion des maladies, etc. Les agences de santé des pays développés comme l'UE, les US, le Canada et l'Australie ont défini des réglementations concernant les SaMDs, et certaines ont déjà élaboré des documents d'orientation tandis que d'autres sont en cours de le faire.
Certains marchés réglementés et non réglementés considèrent les logiciels comme des dispositifs médicaux, mais ne disposent pas de lignes directrices différenciées et spécifiques pour la classification des logiciels en tant que dispositifs médicaux (SaMD). Ils suivent les lignes directrices harmonisées et acceptées au niveau international pour l'évaluation et l'approbation des logiciels.
Voici quelques-unes des principales directives disponibles concernant l'enregistrement des logiciels en tant que dispositif médical (SaMD) :
- Lignes directrices de l'IMDRF pour la classification, le Système de Gestion de la Qualité (SGQ), l'évaluation de la cybersécurité et l'évaluation clinique.
- Le EU MDR 2017/745 a détaillé les exigences réglementaires et les lignes directrices pour cette catégorie de dispositifs.
- Les lignes directrices du MDCG sur la qualification et la classification des SaMD, les exigences des Clinical evaluation reports (CER) / Rapports d'évaluation des performances (PER) pour les SaMD.
- Les directives de la US FDA sur la cybersécurité, l'évaluation clinique et les exigences d'enregistrement pour différents types de logiciels tels que les systèmes d'aide à la décision, les systèmes d'archivage et de communication d'images (PACS), les applications mobiles, etc.
- Document d'orientation de Santé Canada sur la définition et la classification des SaMD.
- Les nouvelles réglementations de la TGA pour les dispositifs médicaux basés sur des logiciels.
L'enregistrement des SaMD sur d'autres marchés mondiaux doit être traité au cas par cas et nécessite une interaction étroite avec l'autorité sanitaire respective pour l'approbation. La procédure générale suivie pour l'enregistrement des SaMD comprend :
- Déterminer si un logiciel donné est qualifié de SaMD.
- Classification des dispositifs en fonction du risque associé.
- Identifier les normes applicables et les exigences en matière de données exigées par l'Agence de Santé concernée.
- Générer des données tel que requis par l'Agence respective.
- Compilation du dossier technique conformément aux exigences du pays.
- Soumission et résolution des questions jusqu'à l'approbation.
- Gestion du cycle de vie après approbation.
Nos compétences
- Services de veille réglementaire (liés au marché et au soutien à l'étiquetage)
- Diligence raisonnable réglementaire / Rapports de stratégie
- Qualification et Classification des SaMD
- Demande de classification de produit au NB
- Analyse des écarts
- Réunions avant soumission avec la FDA
- Identification des normes applicables
- Activités de gestion des risques
- Soutien à la gestion des risques
- Support d'étiquetage
- Création/examen/mise à jour des procédures/modèles spécifiques aux SaMD
- UDI/GUDID
- Enregistrement du produit (Enregistrement du logiciel)
- Enregistrement d'établissement
- Enregistrement des dispositifs
- Réponse aux requêtes des autorités compétentes — services SaMD
Pourquoi Freyr ?
Foire aux questions (FAQ)
La réglementation des logiciels médicaux est supervisée par divers organismes de réglementation mondiaux, notamment la FDA aux US, l'EMA en Europe et la PMDA au Japon. Ces agences classent les logiciels médicaux en fonction du risque et établissent des lignes directrices pour la sécurité, la qualité et l'efficacité. La conformité aux normes ISO, telles que ISO 13485 et 62304, est requise.
La détermination de la classification des risques des logiciels en tant que dispositif médical (SaMD) implique l'évaluation de facteurs tels que l'usage prévu et le préjudice potentiel. Les SaMD sont classés comme les dispositifs médicaux traditionnels en fonction de l'importance des informations fournies pour les décisions de santé et de l'état de la situation ou de la condition de santé comme non grave, grave et critique. Les lignes directrices réglementaires et la consultation d'experts sont cruciales dans ce processus, garantissant la conformité et la sécurité des patients.
Le SaMD désigne un logiciel destiné à être utilisé à une ou plusieurs fins médicales, sans faire partie d'un dispositif médical physique. Il fonctionne sur des plateformes informatiques à usage général telles que les smartphones, les tablettes ou les ordinateurs personnels. D'autre part, le SiMD est un logiciel qui est un composant intégral d'un dispositif médical physique, contribuant à sa fonctionnalité et à ses performances. Le SiMD ne peut pas être utilisé indépendamment et dépend du dispositif médical associé pour remplir sa fonction prévue.
Un logiciel intégré à un dispositif médical matériel et nécessaire pour atteindre l'objectif médical visé N'EST PAS considéré comme un SaMD.
Le calendrier pour atteindre la conformité des SaMD est influencé par la classe de risque et les exigences réglementaires. Mais, avec l'assistance réglementaire appropriée, vous pouvez assurer un processus de conformité plus fluide avec un minimum de risques.
Enregistrement des dispositifs médicaux
- Stratégie réglementaire complète pour les SaMDs.
- Soutien en veille réglementaire et de marché.
- Services de classification et d'enregistrement des produits pour les SaMDs.
- Soutien réglementaire pour les documents de développement de produits SaMD.
- Services de consultation sur les études d'évaluation clinique des SaMD.
- Gestion des modifications post-approbation.
- Service de représentation locale.

- Stratégie réglementaire complète pour les SaMDs.
- Soutien en veille réglementaire et de marché.
- Services de classification et d'enregistrement des produits pour les SaMDs.
- Soutien réglementaire pour les documents de développement de produits SaMD.
- Services de consultation sur les études d'évaluation clinique des SaMD.
- Gestion des modifications post-approbation.
- Service de représentation locale.