

Aperçu de l'enregistrement des dispositifs médicaux en Nouvelle-Zélande
En Nouvelle-Zélande, les dispositifs médicaux sont réglementés par la New Zealand Medicines and Medical Devices Safety Authority (Medsafe) conformément au Medicines Regulations 1984, au Medicines Act 1981 et au Medicines (Database of Medical Devices) Regulations 2003. Bien qu'une approbation préalable à la mise sur le marché ne soit pas nécessaire, l'enregistrement des produits dans la base de données du système Electronic Web Assisted Notification of Devices (WAND) dans les 30 jours suivant le lancement commercial est obligatoire. Des documents prouvant la sécurité et l'efficacité, tels qu'une certification d'organismes reconnus comme un organisme notifié de l'UE ou Santé Canada, peuvent être demandés par Medsafe.
L'équipe d'experts en réglementation des dispositifs médicaux de Freyr apporte une expérience considérable pour guider les entreprises de dispositifs médicaux tout au long du processus d'enregistrement Medsafe en Nouvelle-Zélande.
![]()
Autorité réglementaire : Autorité de sécurité des dispositifs médicaux (Medsafe)![]()
Réglementation :Le Règlement sur les médicaments (Base de données des dispositifs médicaux), 2003
Loi sur les médicaments de 1981
Règlement sur les médicaments de 1984![]()
Voie réglementaire : Système électronique de notification des dispositifs assisté par le Web (WAND)![]()
Représentant autorisé : Promoteur de dispositif médical![]()
Exigence SMQ : Certification ISO 13485:2016![]()
Évaluation des données techniques : Autorité de sécurité des dispositifs médicaux (Medsafe)![]()
Validité de la licence : Les enregistrements de dispositifs en Nouvelle-Zélande n'expirent pas. Les dispositifs jugés présenter une menace majeure pour le public peuvent être retirés du marché.![]()
Exigences d'étiquetage : Article 12, paragraphe 4, du Règlement sur les médicaments de 1984 et GHTF/SG1/N43:2005![]()
Format de soumission : Système électronique de notification des dispositifs assisté par le Web (WAND)![]()
Langue : Anglais
Classification des dispositifs médicaux en Nouvelle-Zélande
Les dispositifs médicaux en Nouvelle-Zélande sont classés par risque en classes I, IIa, IIb, III et AIMD, conformément aux critères de l'International Medical Device Regulators Forum (IMDRF). Cette classification influe sur le niveau de contrôle réglementaire nécessaire. La classification est basée sur des caractéristiques telles que l'objectif prévu du dispositif, la durée de contact avec le corps, l'invasivité et son caractère actif ou inactif. Les dispositifs de classe supérieure sont soumis à une surveillance réglementaire plus stricte. Medsafe est l'entité réglementaire en Nouvelle-Zélande qui supervise ces classifications et réglementations.
| Classification Medsafe des dispositifs médicaux (autres que les DIV) | Risque |
|---|---|
| Classe I de base | Faible risque |
| Classe I avec fonction de mesurage | Faible risque |
| Classe I stérile | Faible risque |
| Classe IIa | Risque faible à moyen |
| Classe IIb | Risque moyen-élevé |
| Classe III et dispositif médical implantable actif (AIMD) | Risque élevé |
| Classification Medsafe des DIV | Risque |
|---|---|
| Depuis juillet 2014, Medsafe ne reconnaît aucun système de classification des risques pour les DIV. Tous les DIV notifiés à WAND doivent utiliser le code de classification des risques du DIV. Le Directeur général de la Santé a autorisé l'exemption pour les DIV conformément à l'annexe 1, paragraphe (i) du Règlement de 2003 sur les médicaments (Base de données des dispositifs médicaux). Mais les fournisseurs de DIV peuvent volontairement notifier leurs dispositifs à la base de données. | |
Représentant autorisé de dispositifs médicaux / Promoteur
Le Représentant Autorisé est désigné comme le Sponsor et agit comme intermédiaire entre le fabricant et Medsafe. Les Sponsors servent de représentants réglementaires pour les produits commercialisés en Nouvelle-Zélande, soumettant les demandes WAND, et agissant comme point de contact principal entre le fabricant et Medsafe pour toutes les questions liées au produit. De plus, Medsafe tient le Sponsor responsable des efforts de vigilance.
Enregistrement des dispositifs médicaux en Nouvelle-Zélande
Enregistrement des dispositifs médicaux, la procédure en Nouvelle-Zélande et la procédure d'inscription WAND en Nouvelle-Zélande varient selon la classe du dispositif.
Dispositifs de classe I- Une déclaration de conformité du fabricant est requise pour les équipements de classe I non stériles et non mesurables ; cependant, elle est rarement déposée auprès d'un organisme de réglementation. Au lieu de cela, le promoteur (ou fournisseur) doit saisir les détails du dispositif dans la base de données WAND (Web Assisted Notification of Devices) dans le cadre du processus de notification Medsafe.
Autres dispositifs de classe
En Nouvelle-Zélande, les sponsors ou fournisseurs sont chargés de s'assurer que les dispositifs médicaux respectent des normes comme l'ISO 13485:2016. La soumission directe d'une déclaration de conformité, d'une certification de système de management de la qualité (SMQ) ou de preuves de fabrication à Medsafe n'est généralement pas requise. Cependant, la conservation de cette documentation est essentielle pour prouver la conformité sur demande.
Medsafe privilégie la surveillance post-commercialisation par rapport à une approbation pré-commercialisation détaillée pour les dispositifs médicaux. Bien que les audits ne soient pas systématiquement menés pendant la phase de notification, Medsafe peut les initier pour les dispositifs à risque plus élevé ou suite à des activités de vigilance et des rapports d'événements indésirables, garantissant ainsi une sécurité et une conformité continues.
Une fois qu'un dispositif est notifié via la base de données WAND, il peut être commercialisé en Nouvelle-Zélande, à condition que le fournisseur respecte constamment les réglementations de Medsafe. Cela exige une conformité continue, en particulier en ce qui concerne la surveillance après commercialisation et les normes de déclaration d'incidents. Les experts en dispositifs médicaux de Freyr soutiennent les services liés à la navigation de ces exigences réglementaires, garantissant que les entreprises maintiennent leur conformité tout au long du cycle de vie du produit.
Flux de processus
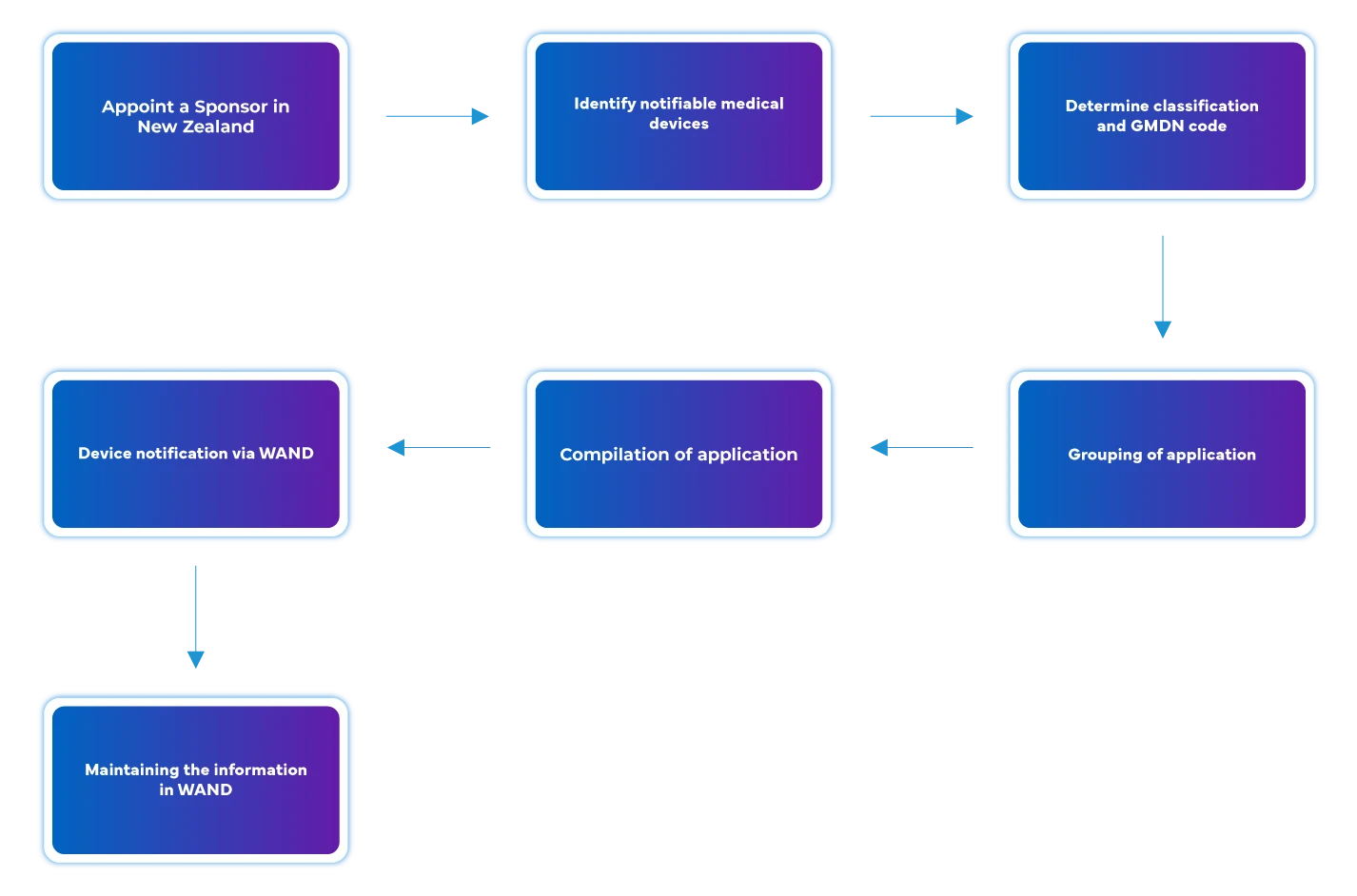
Gestion du cycle de vie des dispositifs post-approbation
Freyr accompagne les fabricants étrangers dans la gestion End-to-End du cycle de vie des dispositifs médicaux, y compris les activités post-approbation en informant les autorités néo-zélandaises via WAND, telles que –
- Gestion des changements post-approbation – modifications aux approbations existantes de dispositifs médicaux, telles que l'ajout de nouvelles variantes, d'accessoires ; l'ajout de nouvelles indications d'utilisation, entre autres.
- Maintien des approbations et des enregistrements.
Doté d'une équipe de professionnels de la réglementation, Freyr offre un soutien complet aux fabricants afin de maintenir les normes de qualité et de sécurité requises pour l'approbation de mise sur le marché. Les spécialistes en veille réglementaire de l'entreprise surveillent méticuleusement les mises à jour des réglementations, s'assurant que les clients sont bien informés des actions nécessaires pour maintenir la conformité de leurs produits aux normes actuelles.
Résumé
| Risque | Classe de dispositif | Audit du SGC | Voie réglementaire | Délais Medsafe | Validité de l'enregistrement (années) |
|---|---|---|---|---|---|
| Faible risque | Classe I de base | Conformité ISO 13485:2016 Remarque : Medsafe n'exige pas d'audits du système de management de la qualité (SMQ), mais recommande fortement de suivre la norme ISO 13485:2016 pour la qualité et la sécurité. Medsafe a l'autorité de mener des audits SMQ pour toute classe de dispositif si des préoccupations de sécurité ou de qualité surviennent. | Enregistrement WAND (Notification) | 1 semaine |
Pas de dates de péremption |
| Faible risque | Classe I de mesure | Enregistrement WAND (Notification) | |||
| Faible risque | Classe I stérile | Enregistrement WAND (Notification) | |||
| Risque faible à moyen | Classe IIa | Enregistrement WAND (Notification) | |||
| Risque moyen-élevé | Classe IIb | Enregistrement WAND (Notification) | |||
| Risque élevé | Classe III | Enregistrement WAND (Notification) |
Remarque : Conformément à la législation actuelle, les enregistrements de dispositifs en Nouvelle-Zélande n'expirent pas, mais les dispositifs considérés comme présentant un risque inacceptable pour le public peuvent être retirés du marché. Cependant, la législation actuelle pourrait être révisée d'ici 2026/2027.
L'expertise de Freyr
- Soutien complet à l'enregistrement des dispositifs médicaux.
- Support LR
- Enregistrement WAND
- Support d'étiquetage
- Gestion des changements post-approbation
- Transfert de licence
- Services de soumission et de liaison avec WAND