
3 min de lectura
En el cambiante mundo de los dispositivos médicos, el cumplimiento normativo no es una tarea puntual, sino un compromiso continuo. La supervisión y actualización continuas de informes clave, como los informes de evaluación clínica (CER), los informes de evaluación del rendimiento (PER) y los informes periódicos de seguridad (PSUR), son fundamentales a lo largo de todo el ciclo de vida de un Dispositivos Médicos, desde la investigación inicial hasta la vigilancia posterior a la comercialización. A medida que el panorama de los avances médicos y los requisitos normativos evolucionan continuamente, garantizar la seguridad y el cumplimiento normativo de los dispositivos médicos y los IVD mediante una redacción médica eficaz sigue siendo la piedra angular del éxito y la viabilidad a largo plazo.
Veamos cómo la gestión del ciclo de vida sigue siendo esencial para el éxito Dispositivos Médicos
El lanzamiento de un Dispositivos Médicos la culminación de años de esfuerzo dedicado a varias fases, como la investigación, el desarrollo, los ensayos clínicos, las presentaciones reglamentarias y la vigilancia posterior a la comercialización. Este proceso se prolonga durante muchos años y en cada fase se acumulan grandes cantidades de datos esenciales, que deben recopilarse y analizarse cuidadosamente para garantizar que el dispositivo siga siendo seguro.
El cumplimiento de la normativa no es cosa de una sola vez, y una gestión eficaz del ciclo de vida ayuda a evitar retrasos que pueden resultar costosos más adelante. Estos retrasos suelen deberse a una planificación ineficaz de cambios imprevistos en la normativa o a incumplimientos que pueden haberse pasado por alto. Las actualizaciones periódicas de documentos críticos como los CER, PER y PSUR garantizan que los fabricantes cumplan los requisitos normativos en constante evolución y mantengan la seguridad de los productos. La gestión del ciclo de vida garantiza que los fabricantes, desde el principio, han planificado cada etapa, por lo que pueden evitar cualquier escollo y garantizar el éxito de la entrada en el mercado y la longevidad de sus dispositivos.
¿Está al día del cumplimiento de la normativa?
Desde vendajes hasta implantes, la Dispositivos Médicos es un sector creativo y de vanguardia con un gran potencial. A pesar del firme deseo de los fabricantes de ofrecer al mercado productos seguros y de alta calidad, sigue existiendo cierta ambigüedad. EU MDR la Dispositivos Médicos (MDD) y EU MDR la Directiva sobre productos sanitarios implantables activos (AIMDD). Esta normativa se implementó para imponer controles más rigurosos, mejorar la seguridad de los pacientes y promover una mayor transparencia en el sector sanitario. Existe mucha incertidumbre sobre estas normas y, con frecuencia, se pasan por alto cuestiones cruciales relacionadas con el cumplimiento. Sin embargo, Dispositivos Médicos deben tomar medidas para evitar dificultades normativas.
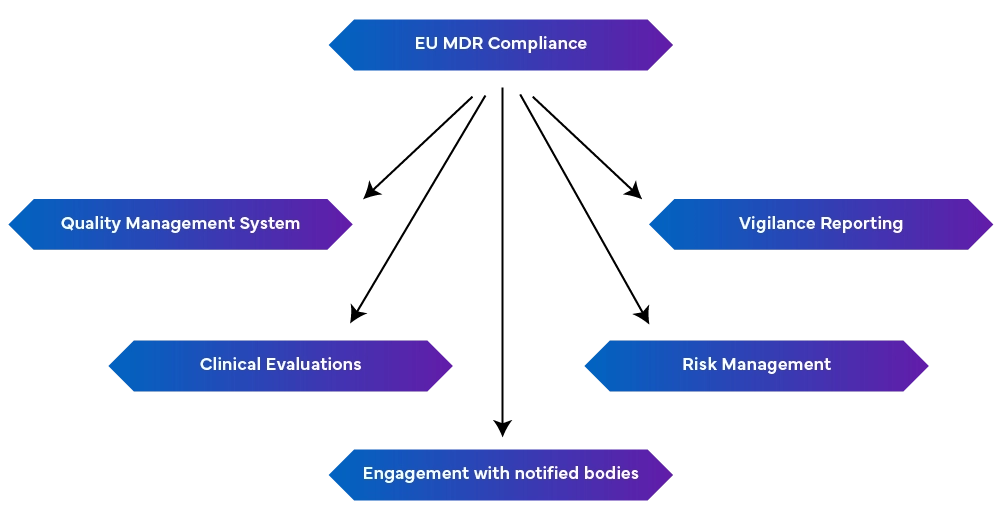
Analicemos los retos más comunes a los que se enfrentan Dispositivos Médicos
- Mantenimiento de CAPA (Corrective and Preventive Action)
- Cumplimiento de los procedimientos de reclamación
- Cumplimiento de los procedimientos de vigilancia
- Evaluación clínica y vigilancia postcomercialización
- Conexión con los organismos notificados
Buenas prácticas para cumplir la normativa
Para superar estos retos, en Freyr hemos creado
-unsólido sistema de gestión de la calidad (EN ISO 13485:2016): Incluye aspectos de producción, como el cumplimiento de la normativa, la documentación técnica, las declaraciones de conformidad de la UE y la gestión de riesgos.
-Informes de vigilancia: Mantener los informes de vigilancia como un proceso continuo y no como un esfuerzo aislado.
- Gestión proactiva de riesgos: Identifica y mitiga los riesgos a lo largo del ciclo de vida del dispositivo, actualizándose periódicamente para abordar los riesgos emergentes.
- Realizar evaluaciones clínicas y vigilancia postcomercialización: Esto se hace demostrando la seguridad y el rendimiento del dispositivo utilizando datos clínicos. Si es necesario, deben realizarse investigaciones clínicas, y las evaluaciones clínicas deben actualizarse periódicamente con datos de vigilancia posterior a la comercialización. También se requieren informes periódicos de actualización de la seguridad que resuman los hallazgos.
- Conexión con los organismos notificados: Los cambios en la normativa pueden afectar a los requisitos del MDR, por lo que es crucial mantener una buena relación con los Organismos Notificados para conseguir actualizaciones y modificaciones rápidas.
Cómo puede ayudarle un experto en normativa
Mantenerse al día con un panorama normativo en constante cambio puede resultar difícil. ¿Por qué no dejar que un experto le guíe a través de este laberinto? Gestionar las exigencias normativas del ciclo de vida Dispositivos Médicosno solo es complejo, sino que también requiere muchos recursos. En el caso de las empresas más pequeñas, donde los recursos internos pueden utilizarse mejor en otras áreas, la ayuda de un socio externo especializado en normativa puede resultar indispensable. Estos socios ofrecen conocimientos especializados sobre la evolución de la normativa y garantizan que sus informes clave, como los CER, PER y PSUR, se actualicen continuamente y cumplan con la normativa.
Puede evitar retrasos costosos, prevenir incumplimientos y agilizar el proceso de aprobación. Además, un experto en reglamentación puede ofrecer soluciones a medida basadas en las necesidades específicas de su producto, garantizando que la vigilancia posterior a la comercialización, el análisis de deficiencias y toda la documentación de conformidad estén actualizados. Su participación puede simplificar todo el proceso, minimizar los riesgos y garantizar que el dispositivo cumpla las expectativas de seguridad y rendimiento.
Freyr puede ayudarle con todas sus necesidades normativas relativas a la gestión del ciclo de vida. Reach en contacto con us hoy mismo.









