
Panorama general de la aprobación previa a la comercialización de Dispositivos Médicos por la USFDA
El proceso de aprobación previa a la comercialización (PMA) de la USFDA es una de las vías de registro de dispositivos proporcionadas por la US FDA, diseñado principalmente para Dispositivos Médicos de Clase III de la FDA. El proceso de aprobación PMA de la FDA para dispositivos de Clase III implica evaluaciones científicas y reglamentarias meticulosas para evaluar la seguridad y eficacia del Dispositivo Médico, asegurando que se cumplan los más altos estándares antes de la autorización de comercialización.
Reserve una reunión con nuestros expertos en aprobación previa a la comercialización
¿Quién debe presentar una Solicitud de Aprobación Previa a la Comercialización (PMA) de Dispositivos Médicos ante la USFDA?
Los fabricantes de Dispositivos Médicos deben presentar una solicitud de PMA si el Dispositivo Médico:
- Es novedoso.
- Pertenece a una clase de alto riesgo.
- No se encuentra en la Base de Datos de Clasificación de Productos.
- No es sustancialmente equivalente (NSE) a dispositivos de Clase I, II o III.
Obtenga asesoramiento experto sobre su solicitud de aprobación previa a la comercialización
¿Cuál es la diferencia entre las solicitudes 510(k), PMA y De-Novo?
Aprobación de Precomercialización
- Dispositivo Médico de Clase III que sustenta la vida humana o que presenta un riesgo potencial e irrazonable de enfermedad o lesión.
- El proceso de aprobación PMA de la FDA requiere ensayos clínicos.
- Requiere una inspección in situ antes de emitir la aprobación de la PMA.
- 180 días naturales
Clasificación De Novo
- Dispositivos novedosos de Clase I y II que no tienen un dispositivo precedente válido.
- Requiere datos de estudios clínicos.
- No se requiere auditoría in situ antes de la aprobación De-Novo.
- 150 días naturales.
Registro 510(k)
- Dispositivos de Clase III de la FDA que tienen equivalencia sustancial con el dispositivo de referencia.
- No requiere pruebas en humanos.
- No se requiere auditoría in situ antes de la autorización 510(k).
- 90 días naturales.
¿Cuáles son los diferentes métodos de solicitud de aprobación previa a la comercialización de la FDA?
Los fabricantes pueden optar por cualquiera de los siguientes cuatro (04) métodos de solicitud PMA que mejor se adapten a su dispositivo:
- PMA Tradicional
- PMA Modular
- Protocolo de Desarrollo de Productos
- Exención para Dispositivos Médicos de Uso Humanitario
¿Cuáles son los requisitos de datos para la aprobación previa a la comercialización de Dispositivos Médicos?
Según 21 CFR parte 814, los solicitantes deben presentar un formulario de solicitud CDRH debidamente cumplimentado, los compromisos requeridos y un expediente técnico de PMA bien redactado a la FDA de US. El expediente técnico deberá incluir los datos no clínicos y clínicos.
Datos No Clínicos – Consiste en datos sobre microbiología, toxicología, inmunología, biocompatibilidad, estrés, desgaste, vida útil y otras pruebas de laboratorio o en animales.
Datos Clínicos – Consiste en datos sobre protocolos de estudio, datos de seguridad y eficacia, reacciones adversas y complicaciones, fallos y reemplazos de dispositivos, información del paciente, quejas del paciente, tabulaciones de datos de todos los sujetos individuales, resultados de análisis estadísticos y cualquier otra información de las investigaciones clínicas.
¿Qué es el Proceso de Solicitud de PMA?
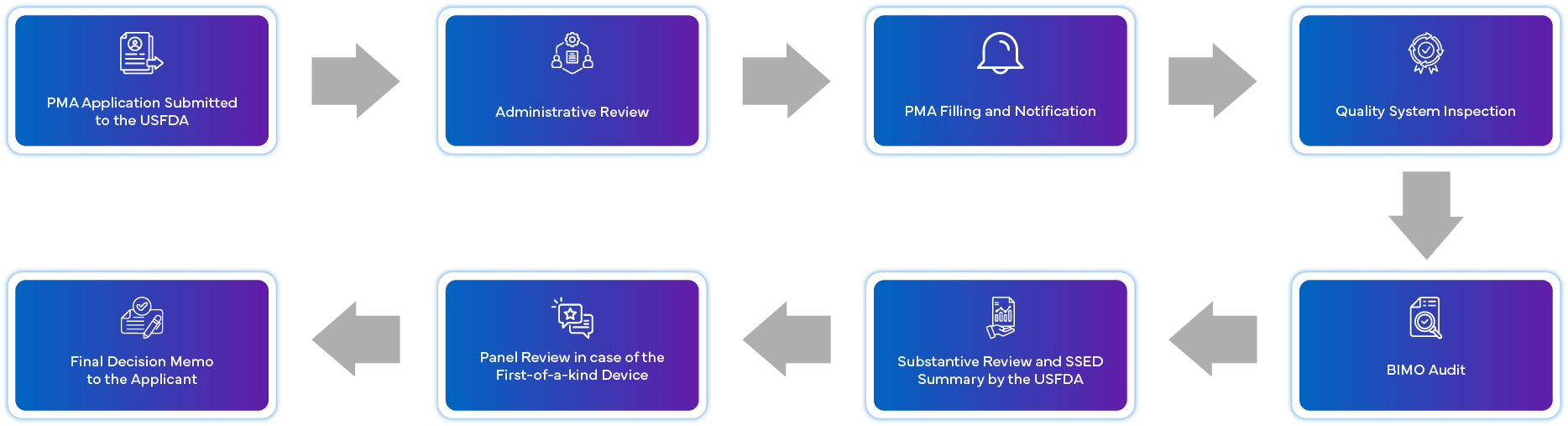
¿Cuáles son los requisitos de cumplimiento post-aprobación para la PMA?
Los dispositivos aprobados bajo la vía PMA deberán cumplir con los requisitos de post-comercialización establecidos por la USFDA. El dispositivo debe cumplir con lo siguiente:
- Requisitos posteriores a la aprobación impuestos en la orden de aprobación de PMA de la FDA.
- Gestión de cambios post-aprobación mediante la presentación oportuna de los suplementos de PMA relevantes
- Presentación de informes post-aprobación (anuales)
- Regulaciones 21 CFR 803 para la Notificación de Dispositivos Médicos (MDR)
- Estudios de vigilancia poscomercialización según lo exigido por la USFDA en las órdenes de aprobación de PMA.
¿Cuáles son las tarifas de la USFDA para revisar la solicitud PMA?
Las tarifas de usuario de MDUFA para la PMA original y los suplementos son las siguientes:
| Tipo de solicitud | Tasas para el año fiscal 2023 (1º de octubre de 2022 hasta el 30º de septiembre de 2023) | |
|---|---|---|
| Tarifa estándar | Tarifa para pequeñas empresas | |
| PMA, PDP, PMR, BLA | $441,547 |
|
| Suplemento Panel-Track | $353,238 | $88,309 |
| Suplemento de 180 días | $66,232 | $16,558 |
| Tarifa Anual para la Notificación Periódica de un dispositivo de Clase III (PMAs, PDPs y PMRs) | $15,454 | $3,864 |
| Aviso de 30 días | $7,065 | $3,532 |
| Suplemento en Tiempo Real | $30,908 | $7,727 |
Con experiencia en el manejo de presentaciones PMA, Freyr puede ayudar a identificar y compilar la información y asistir en la preparación y revisión de la solicitud.
Experiencia y ventajas en la aprobación previa a la comercialización de Dispositivos Médicos por la USFDA
- Debida Diligencia Reglamentaria
- Cumplimiento de la Inspección del Sistema de Calidad
- Cumplimiento de auditoría BIMO
- Compilación del expediente técnico de la PMA
- Publicación y Creación de eCopy
- Validación y presentación de eCopy
- Aborda la respuesta RTA y las deficiencias
- Servicios de enlace hasta la aprobación previa a la comercialización de la FDA.
- Asesoramiento sobre deficiencias
- Registro de Dispositivos y Establecimientos
- Gestión de Suplementos de PMA y avisos de 30 días
- Presentaciones de Informes Periódicos Anuales
- Auditorías simuladas y formación sobre 21 CFR 820

- Experiencia en la gestión de muchas presentaciones de PMA a la FDA para diversas categorías de dispositivos.
- Equipo experto para la solicitud de aprobación previa a la comercialización de la FDA según los requisitos reglamentarios.
- Soporte adicional para gestionar consultas relacionadas con la PMA.
- Presentación a tiempo de los entregables
- Al día con las nuevas enmiendas de la US FDA
